



At the end of their normal life span (approximately 120 days), senescent red blood cells (RBCs) are removed from the circulation. Hemolysis is defined as premature destruction and hence a shortened RBC life span (< 120 days). Anemia results when bone marrow production can no longer compensate for the shortened RBC survival; this condition is termed uncompensated hemolytic anemia. If the bone marrow can compensate, the condition is termed compensated hemolytic anemia.
Etiology of Hemolytic Anemia
Hemolysis can be classified according to whether the hemolysis is:
Extrinsic: From a source outside the red cell; disorders extrinsic to the RBC are usually acquired
Intrinsic: Due to an defect within the red cell; intrinsic RBC abnormalities (see table ) are usually inherited
Disorders extrinsic to the red blood cell
Causes of disorders extrinsic to the RBC include:
Medications (drug-induced immune hemolytic anemia eg, quinine, quinidine, beta-lactam antibiotics, methyldopa, sulfamethoxazole/trimethoprim, fludarabine), oxidants (oxidant hemolysis eg, dapsone), and intravenous immunoglobulin
Immunologic abnormalities (eg, autoimmune hemolytic anemia, acquired thrombotic thrombocytopenic purpura, transfusion reactions)
Infections
Mechanical intravascular hemolysis (microangiopathic hemolytic anemia, including thrombotic microangiopathies, prosthetic heart valve, march hemoglobinuria)
Toxins (eg, lead, copper, insect bites/venom)
Thermal injury
Infectious organisms may cause hemolytic anemia through the following mechanisms:
Direct action of toxins (eg, Clostridium perfringens)
Invasion and destruction of the RBC by the organism (eg, Plasmodium species, Bartonella species, Babesia species)
Antibody production (eg, Epstein-Barr virus, mycoplasma)
Hemolytic-uremic syndrome (caused by Shiga toxin–producing Escherichia coli)
Intrinsic red blood cell abnormalities
Defects intrinsic to the RBC that can cause hemolysis involve abnormalities of the following:
RBC membrane
Cell metabolism
Hemoglobin structure
Abnormalities include:
Hereditary cell membrane disorders (eg, hereditary spherocytosis due to quantitative and functional abnormalities of certain RBC membrane proteins [alpha- and beta-spectrin, protein 4.2, Band 3, ankyrin])
Acquired cell membrane disorders (eg, paroxysmal nocturnal hemoglobinuria)
Disorders of RBC metabolism (eg, glucose-6-phosphate dehydrogenase (G6PD) deficiency)
Hemoglobinopathies (eg, sickle cell disease, thalassemias, unstable hemoglobins—ie, rare, inherited variants affecting globin genes that decrease stability and solubility of hemoglobin)
Hemolytic Anemias
Mechanism | Disorder or Agent |
|---|---|
Disorders extrinsic to the red blood cell | |
Immunologic abnormalities | Autoimmune hemolytic anemias:
|
Infectious organisms (direct invasion) | Babesia Bartonella bacilliformis Plasmodium falciparum Plasmodium malariae Plasmodium vivax |
Mechanical trauma | Disseminated intravascular coagulation (DIC) Extracorporeal membrane oxygenation (ECMO) March hemoglobinuria Valvular heart disorders and mechanical valves Ventricular assist devices Primary thrombotic microangiopathies (TMA), including thrombotic thrombocytopenic purpura, atypical hemolytic uremic syndrome (complement-mediated TMA), hemolytic uremic syndrome (Shiga toxin-mediated) Secondary TMAs (ie, disorders manifesting with microangiopathic hemolytic anemia and thrombocytopenia as well as organ damage), including transplant-associated TMA, HELLP (Hemolysis, Elevated Liver Enzymes, and Low Platelets) syndrome/preeclampsia with severe features, autoimmune TMA (eg, associated with systemic lupus erythematosus, catastrophic antiphospholipid syndrome, scleroderma renal crisis), drug-Induced TMA, or related to severe hypertension, infection, or malignancy |
Reticuloendothelial hyperactivity | |
Toxin production by infectious organisms | Clostridium perfringens Shiga toxin |
Toxins | Compounds with oxidant potential (eg, dapsone, phenazopyridine, naphthalene), well water Copper (Wilson disease) Lead Insect venom Snake venom |
Thermal injury | Associated with burns |
Intrinsic red blood cell abnormalities | |
Acquired red blood cell membrane disorders | Severe hypophosphatemia (depletion of adenosine triphosphate [ATP]) Paroxysmal nocturnal hemoglobinuria Stomatocytosis Spur cell anemia (acanthocytosis) |
Congenital red blood cell membrane disorders | |
Disorders of hemoglobin synthesis | Unstable hemoglobins |
Disorders of red blood cell metabolism | Glycolytic pathway defects (eg, pyruvate kinase deficiency) Hexose monophosphate shunt defects (eg, glucose-6-phosphate dehydrogenase [G6PD] deficiency) |
Pathophysiology of Hemolytic Anemia
Hemolysis may be:
Acute
Chronic
Episodic
Hemolysis may also be:
Extravascular
Intravascular
Both
Normal red blood cell processing
Senescent RBCs lose membrane and are cleared from the circulation by the phagocytic cells of the reticuloendothelial system in the spleen, liver, and bone marrow. Hemoglobin is broken down in these cells primarily by the heme oxygenase system. The iron is conserved and reutilized, and heme is degraded to bilirubin, which is conjugated in the liver to bilirubin glucuronide and excreted in the bile.
Extravascular hemolysis
Most pathologic hemolysis is extravascular and occurs when damaged or abnormal RBCs are cleared from the circulation by the spleen and liver. The spleen contributes to hemolysis by destroying mildly abnormal RBCs or cells coated with warm antibodies. An enlarged spleen may sequester even normal RBCs. Severely abnormal RBCs or RBCs coated with IgG antibodies or complement (C3) are destroyed within the spleen and liver, which (because of its large blood flow) can remove damaged cells efficiently. In extravascular hemolysis, the peripheral smear may show spherocytes or with cold agglutinins, erythrocyte agglutination if the blood is not warmed upon collection.
Intravascular hemolysis
Intravascular hemolysis is an important reason for premature RBC destruction and occurs when the cell membrane has been severely damaged by any of a number of different mechanisms, including the following:
Immune phenomena
Direct trauma (eg, march hemoglobinuria)
Shear stress (eg, defective mechanical heart valves, thrombotic microangiopathies [TMA])
Toxins (eg, clostridial toxins, venomous snake bite)
Intravascular hemolysis results in hemoglobinemia when the amount of hemoglobin released into plasma exceeds the hemoglobin-binding capacity of the plasma-binding protein haptoglobin, a protein normally present in concentrations of approximately 100 mg/dL (1.0 g/L) in plasma. Thus, intravascular hemolysis reduces unbound plasma haptoglobin. With hemoglobinemia, unbound hemoglobin dimers are filtered into the urine and reabsorbed by renal tubular cells; hemoglobinuria results when reabsorptive capacity is exceeded. Iron is released from catabolized hemoglobin and embedded in hemosiderin within the tubular cells; some of the iron is assimilated for reutilization and some reaches the urine when the tubular cells slough.
Consequences of hemolysis
Unconjugated (indirect) hyperbilirubinemia and jaundice occur when the conversion of hemoglobin to bilirubin exceeds the liver’s capacity to conjugate and excrete bilirubin. Bilirubin catabolism causes increased stercobilin in the stool and urobilinogen in the urine and sometimes cholelithiasis.
Increased production of erythropoietin by the kidneys in response to the ensuing anemia causes the bone marrow to accelerate production and release of RBCs, resulting in reticulocytosis.
Symptoms and Signs of Hemolytic Anemia
Systemic manifestations of hemolytic anemias resemble those of other anemias and include pallor, fatigue, dizziness, and weakness. Scleral icterus and/or jaundice may occur, and the spleen may enlarge. Thrombosis increases with many forms of hemolytic anemia.
Hemolytic crisis (acute, severe hemolysis) is uncommon; it may be accompanied by chills, fever, back and abdominal pain, and shock. Hemoglobinuria causes red or reddish-brown urine.
Diagnosis of Hemolytic Anemia
Peripheral smear and reticulocyte count
Serum bilirubin (indirect), lactate dehydrogenase (LDH), and haptoglobin
Sometimes direct antiglobulin (Coombs) test and/or hemoglobinopathy screen
Urinalysis
Hemolysis is suspected in patients with anemia and reticulocytosis. If hemolysis is suspected, a peripheral smear is examined and serum bilirubin, LDH, and haptoglobin are measured. The peripheral smear and reticulocyte count are the most important tests to diagnose hemolysis. Antiglobulin testing or hemoglobinopathy screening (eg, high-performance liquid chromatography [HPLC]) can help identify the cause of hemolysis based on the results of these laboratory investigations. However, in some patients with a hemolytic anemia, the reticulocyte count fails to increase due to factors such as renal insufficiency, infection, or bone marrow failure, creating a hematologic emergency. This lack of the expected compensation (reticulocytopenia) necessitates prompt transfusion therapy.

The presence of abundant spherocytes in the peripheral blood smear suggests either hereditary spherocytosis (HS) or autoimmune hemolytic anemia (AIHA). Spherocytes are spherical red blood cells without an area of central pallor and are usually slightly smaller in size than the average red cell. Spherocytes have increased osmotic fragility (because of decreased distensibility associated with reduced surface membrane area) in hypotonic saline, and this test (osmotic fragility test) is positive in HS and in AIHA. The direct antiglobulin (Coombs) test distinguishes between them; the result is positive in AIHA and negative in HS.
By permission of the publisher. From Tefferi A, Li C. In Atlas of Clinical Hematology. Edited by JO Armitage. Philadelphia, Current Medicine, 2004.

Schistocytes (see arrows) are damaged red blood cells, which may occur in microangiopathic hemolytic anemia (including disseminated intravascular coagulation, thrombotic thrombocytopenic purpura/hemolytic-uremic syndrome, and valvular hemolysis).
By permission of the publisher. From Tefferi A, Li C. In Atlas of Clinical Hematology. Edited by JO Armitage. Philadelphia, Current Medicine, 2004.
Abnormalities of RBC morphology often suggest the presence and cause of hemolysis (see table ). The presence of spherocytes on the peripheral smear suggests an extravascular cause of hemolysis such as autoimmune hemolytic anemia or hereditary spherocytosis, while the presence of schistocytes or other fragmented red cells suggests an intravascular cause such as microangiopathic hemolytic anemia (eg, thrombotic thrombocytopenic purpura, hemolytic-uremic syndrome, complement-mediated TMA, or valvular hemolysis). Other suggestive findings include increased levels of serum LDH and indirect bilirubin, decreased haptoglobin, and the presence of urinary urobilinogen.
Haptoglobin levels can decrease because of hepatocellular dysfunction and can increase because of systemic inflammation. Intravascular hemolysis reduces unbound plasma haptoglobin. Intravascular hemolysis is also suggested by urinary hemosiderin. Urinary hemoglobin, like hematuria and myoglobinuria, produces a positive benzidine reaction on dipstick testing; it can be differentiated from hematuria by the absence of RBCs on microscopic urine examination. Free hemoglobin may make plasma reddish brown, noticeable often in centrifuged blood; myoglobin does not.
Once hemolysis has been identified, the etiology is sought. To narrow the differential diagnosis in hemolytic anemias:
Consider risk factors (eg, geographic location, genetics, underlying disorder)
Examine the patient for splenomegaly
Do a direct antiglobulin (direct Coombs) test if cause is not suggested by initial laboratory investigations
Most hemolytic anemias cause abnormalities in one of these variables, and so test results can direct further testing.
Other laboratory tests that can help discern the causes of hemolysis include the following:
Quantitative hemoglobin electrophoresis and HPLC
RBC enzyme assays
Flow cytometry for paroxysmal nocturnal hemoglobinuria (PNH) clone
Cold agglutinins
Osmotic fragility
Genetic testing for membranopathies and enzymopathies
If thrombotic microangiopathies are being considered, ADAMTS13 activity, Shiga toxin testing if diarrhea is present, and consider sequencing of complement regulatory genes and factor H autoantibody testing in cases of suspected complement-mediated TMA, though this testing is not diagnostic and the decision for initial treatment should not be based on this
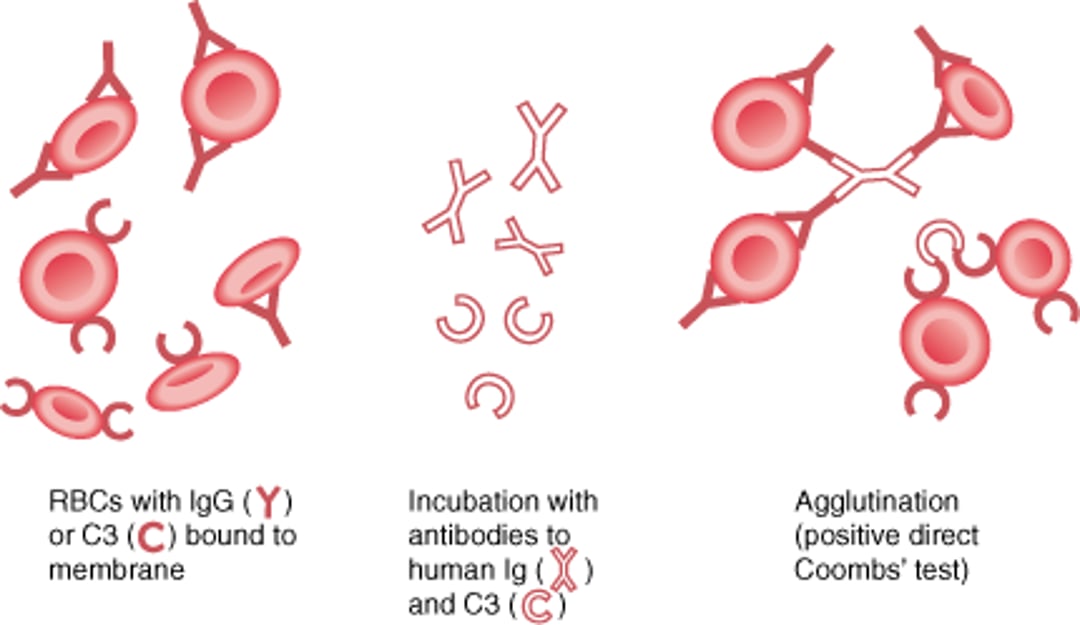
Direct Antiglobulin (Direct Coombs) Test
The direct Coombs test is used to determine whether red blood cell (RBC)-binding antibody (IgG) or complement (C3) is present on RBC membranes. The patient's RBCs are incubated with antibodies to human IgG and C3. If IgG or C3 is bound to RBC membranes, agglutination occurs–a positive result. A positive result suggests the presence of autoantibodies to the patient's RBCs. If the patient has received a transfusion in the last 3 months, a positive result could also represent alloantibodies to transfused RBCs (usually occurring in acute or delayed hemolytic reaction). |

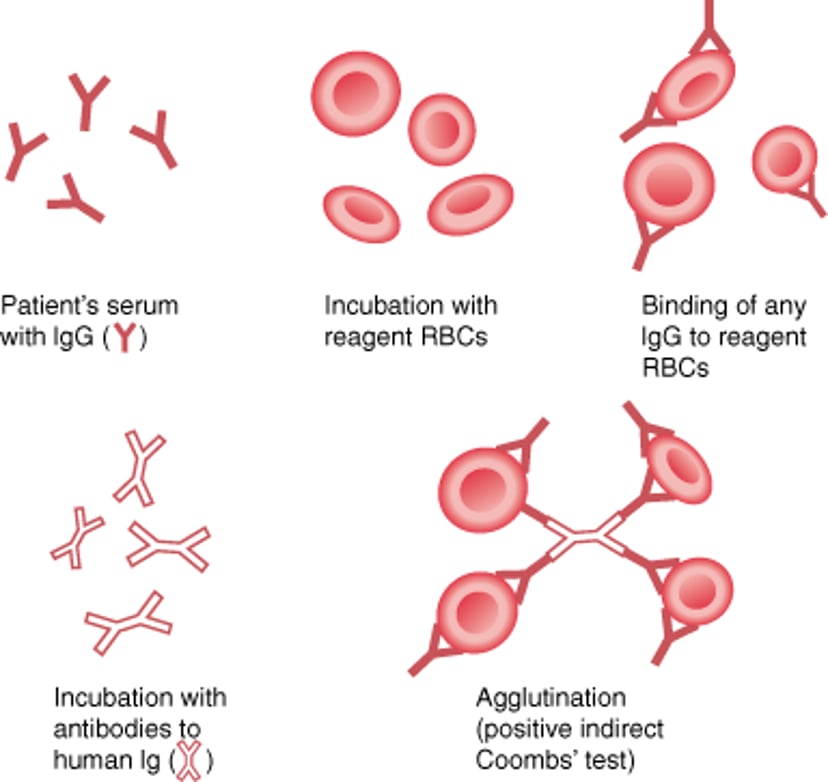
Indirect Antiglobulin (Indirect Coombs) Test
The indirect Coombs test is used to detect IgG antibodies against red blood cells (RBCs) in a patient's serum. The patient's serum is incubated with reagent RBCs; then Coombs serum (antibodies to human IgG, or human anti-IgG) is added. If agglutination occurs, IgG antibodies (autoantibodies or alloantibodies) against RBCs are present. This test is also used to determine the specificity of an alloantibody. |

Red Blood Cell Morphologic Changes in Hemolytic Anemias
RBC Morphology | Causes |
|---|---|
Acanthocytes (spur cells) | Anorexia Liver disease Neuroacanthocytosis |
Agglutinated cells | |
Echinocytes | Liver dysfunction Renal dysfunction |
Heinz bodies (bite or blister cells) | Glucose-6-phosphate dehydrogenase (G6PD) deficiency Oxidant stress Unstable hemoglobin |
Nucleated erythroblasts and basophilic stippling | |
Schistocytes | Microangiopathic hemolytic anemia (MAHA), including disseminated intravascular coagulation (DIC) and thrombotic microangiopathy (TMA) Valve hemolysis, Intravascular prostheses, and extracorporeal devices Vitamin B12 deficiency (pseudo-thrombotic thrombocytopenic purpura [pseudo-TTP]) |
Sickle cells | |
Spherocytes | Transfused blood |
Target cells | Hemoglobinopathies (eg, sickle cell disease, hemoglobin C disease, thalassemias) Liver dysfunction Post-splenectomy |
The American Society of Hematology (ASH) image bank provides images of red blood cell morphologic changes. | |
Treatment of Hemolytic Anemia
Treatment depends on the specific mechanism of hemolysis.
Glucocorticoids are helpful in the initial treatment of warm antibody autoimmune hemolysis. Transfusions are used in patients with symptomatic anemia or when there is reticulocytopenia.
Splenectomy is beneficial in some situations, particularly when splenic sequestration is the major cause of RBC destruction. If possible, splenectomy is delayed until 2 weeks after vaccination with the following:
In cold agglutinin disease, avoidance of cold is recommended, and blood will need to be warmed before transfusion. Folate replacement is needed for patients with ongoing long-term hemolysis.
Drug Information for the Topic





