



Wilson disease (also known as progressive hepatolenticular degeneration) results in accumulation of copper in the liver and other organs. Hepatic or neurologic symptoms develop. Diagnosis is based on a low serum ceruloplasmin level, high urinary excretion of copper, and sometimes liver biopsy results. Treatment consists of a low-copper diet and medications such as penicillamine or trientine.
Wilson disease is a disorder of copper metabolism that affects men and women; approximately 1 person in 30,000 has the disorder. Affected people are homozygous for the autosomal recessive gene, located on chromosome 13. Heterozygous carriers, who constitute approximately 1.1% of the population, are asymptomatic.
Treatment consists of a low-copper diet and medications such as penicillamine or trientine (1, 2).
References
1. Ala A, Walker AP, Ashkan K, Dooley JS, Schilsky ML. Wilson's disease. Lancet. 2007;369(9559):397-408. doi:10.1016/S0140-6736(07)60196-2
2. Roberts EA, Schilsky ML. Current and Emerging Issues in Wilson's Disease. N Engl J Med. 2023;389(10):922-938. doi:10.1056/NEJMra1903585
Pathophysiology of Wilson Disease
The genetic defect in Wilson disease impairs copper transport. The impaired transport decreases copper secretion into the bile, thus causing copper overload and resultant accumulation in the liver, which begins at birth. The impaired transport also interferes with incorporation of copper into the copper protein ceruloplasmin, thus decreasing serum levels of ceruloplasmin.

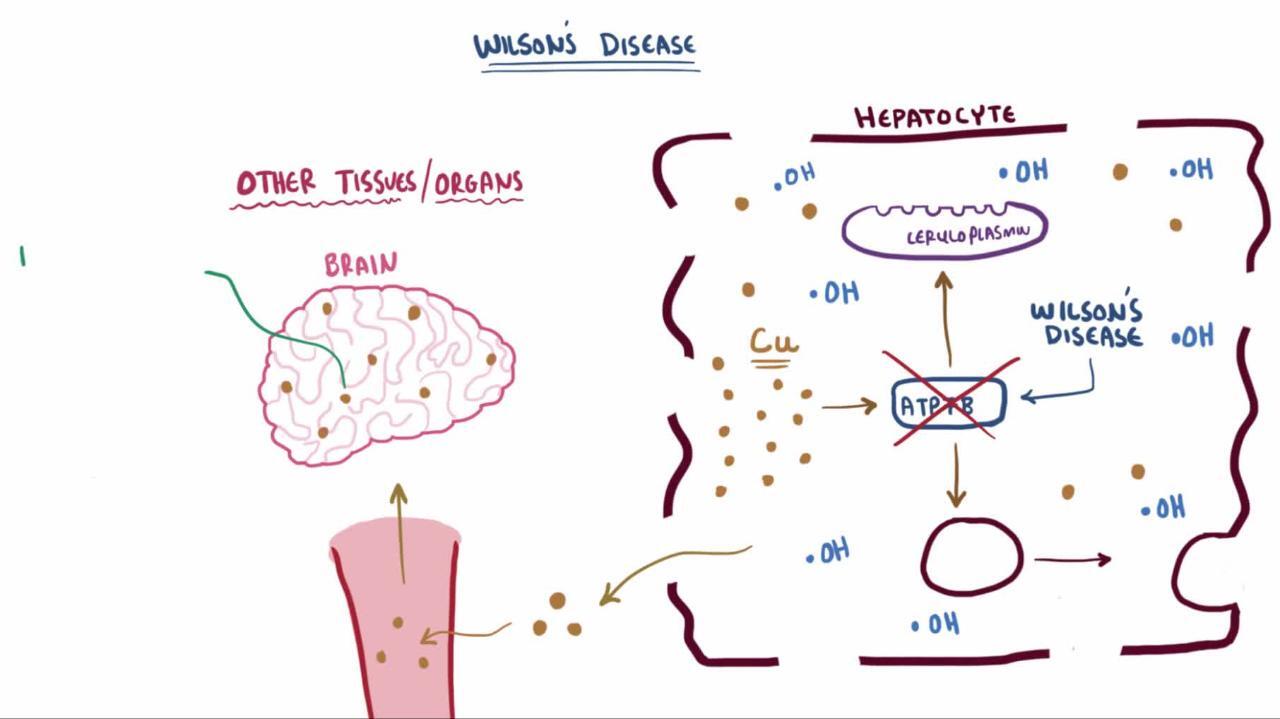
Hepatic fibrosis develops, ultimately causing cirrhosis. Copper diffuses out of the liver into the blood, then into other tissues. It is most destructive to the brain but also damages the kidneys and reproductive organs and causes hemolytic anemia. Some copper is deposited around the rim of the cornea and edge of the iris, causing Kayser-Fleischer rings. The rings appear to encircle the iris.
Symptoms and Signs of Wilson Disease
Symptoms of Wilson disease usually develop between ages 5 and 35 but can develop from age 2 to 72 years.
In almost half of patients, particularly adolescents, the first symptom is
Hepatitis—acute, chronic active, or fulminant
But hepatitis may develop at any time.
In approximately 40% of patients, particularly young adults, the first symptoms reflect
Central nervous system (CNS) involvement
Motor deficits are common, including any combination of tremors, dystonia, dysarthria, dysphagia, chorea, drooling, and incoordination. CNS symptoms may also manifest as cognitive or psychiatric abnormalities.

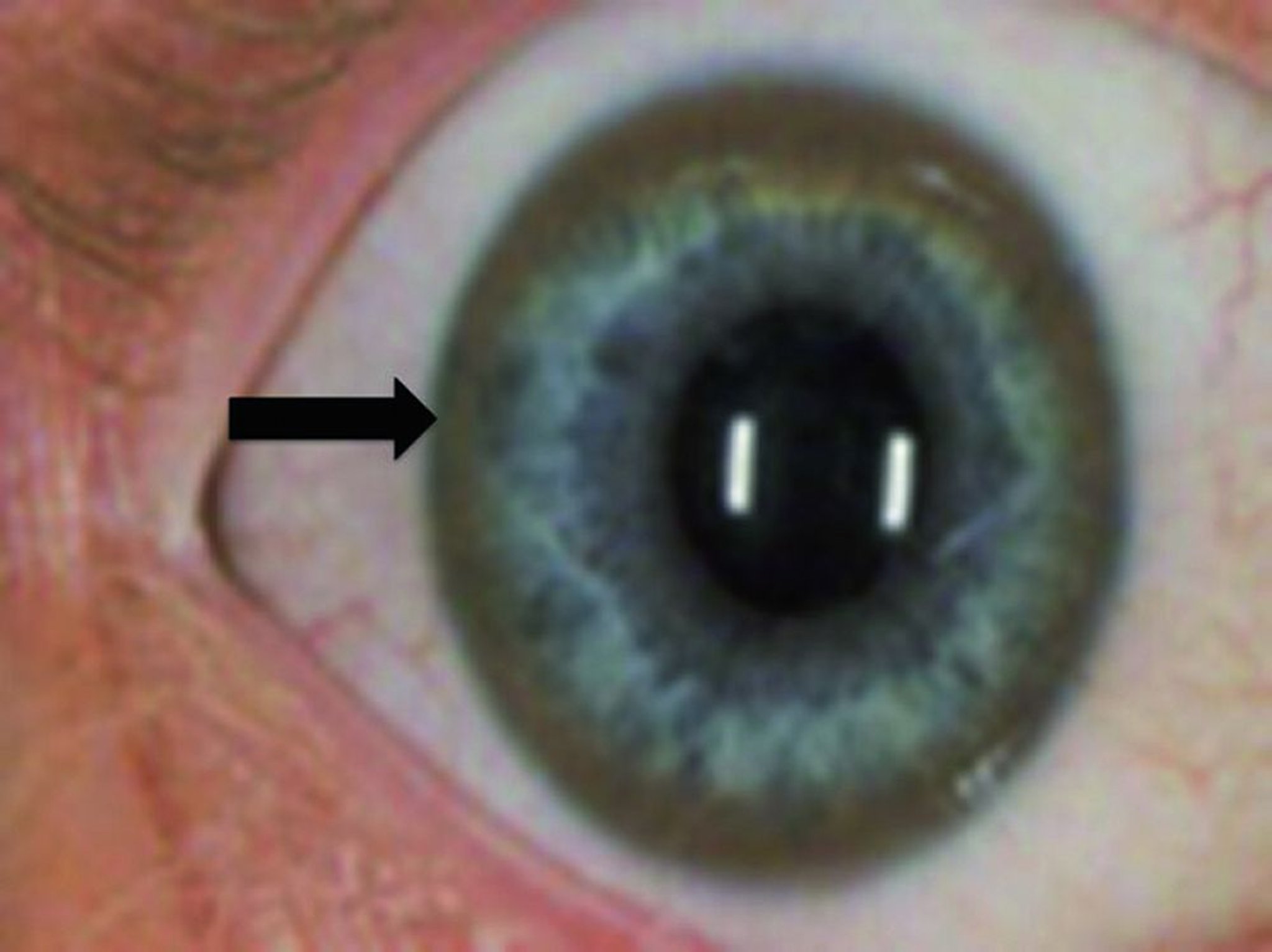
Kayser-Fleischer rings (indicated by the solid black arrow) are gold, greenish gold, golden brown, or brownish green rings in Descemet membrane in the limbic region of the cornea. The rings result from copper deposits in the cornea.
© Springer Science+Business Media
In 5 to 10% of patients, the first symptom is incidentally noted gold or greenish gold Kayser-Fleischer rings or crescents (due to copper deposits in the cornea), amenorrhea or repeated miscarriages, or hematuria.
Diagnosis of Wilson Disease
Slit-lamp examination for Kayser-Fleischer rings
Levels of serum ceruloplasmin, sometimes serum copper, and 24-hour urinary copper excretion
Sometimes confirmation by penicillamine provocation test or liver biopsy
Wilson disease should be suspected in people < 40 with any of the following:
An unexplained hepatic, neurologic, or psychiatric disorder
An unexplained persistent elevation in hepatic transaminases
A sibling, parent, or cousin with Wilson disease
If Wilson disease is suspected, slit-lamp examination for Kayser-Fleischer rings is required, and serum ceruloplasmin levels and 24-hour urinary copper excretion are measured (1). Serum copper levels may be measured, but ceruloplasmin levels are usually sufficient. Transaminase levels are also often measured; high transaminase levels are consistent with the diagnosis.
Kayser-Fleischer rings
These rings plus typical motor neurologic abnormalities or a decrease in ceruloplasmin are nearly pathognomonic for Wilson disease. Rarely, these rings occur in other liver disorders (eg, biliary atresia, primary biliary cirrhosis), but ceruloplasmin levels should be unaffected.
Ceruloplasmin
Serum ceruloplasmin (normally 20 to 35 mg/dL [200 to 350 mg/L]) is usually low in Wilson disease but can be normal. It can also be low in heterozygous carriers and those with other liver disorders (eg, viral hepatitis, drug- or alcohol-induced liver disease). A low ceruloplasmin level in a patient with a Kayser-Fleischer ring is diagnostic. Also, a level < 5 mg/dL (< 50 mg/L) is highly suggestive regardless of clinical findings.
Serum copper
Serum copper levels are sometimes measured; however, they may be high, normal, or low.
Urinary copper excretion
In Wilson disease, 24-hour urinary copper excretion (normally, ≤ 30 mcg/day) is usually > 100 mcg/day. If serum ceruloplasmin is low and urinary copper excretion is high, diagnosis is clear. If levels are equivocal, measuring urinary copper excretion after penicillamine is given (penicillamine provocation test) may confirm the diagnosis; this test is not usually done in adults because cutoff values are not well established.
Liver biopsy
In unclear cases (eg, elevated transaminases, no Kayser-Fleischer rings, indeterminate values for ceruloplasmin and urinary copper), the diagnosis is made by doing a liver biopsy to measure hepatic copper concentration. However, false-negative results may occur because of a sampling error (due to large variations in copper concentrations in the liver) or fulminant hepatitis (causing necrosis that releases large amounts of copper).
Screening for Wilson disease
Because early treatment is most effective, screening is indicated for anyone who has a sibling, cousin, or parent with Wilson disease (1). Screening consists of a slit-lamp examination and measurement of transaminase levels, serum copper and ceruloplasmin, and 24-hour urine copper excretion. If available, screening should include genetic testing for ATP7B mutations. If any results are abnormal, liver biopsy is done to measure hepatic copper concentration.
Infants should not be tested until after age 1 year because ceruloplasmin levels are low during the first few months of life. Children < 6 years with a family history of Wilson disease and normal test results should be retested 5 to 10 years later.
Diagnosis reference
1. Schilsky ML, Roberts EA, Bronstein JM, et al. A multidisciplinary approach to the diagnosis and management of Wilson disease: 2022 Practice Guidance on Wilson disease from the American Association for the Study of Liver Diseases. Hepatology. Published online December 7, 2022. doi:10.1002/hep.32801
Treatment of Wilson Disease
Penicillamine or trientine
Low-copper diet
For maintenance, lifelong low-dose penicillamine or trientine, or oral zinc
Continual, lifelong treatment of Wilson disease is mandatory regardless of whether symptoms are present (1). A low-copper diet (eg, avoiding beef liver, cashews, black-eyed peas, vegetable juice, shellfish, mushrooms, and cocoa) and use of penicillamine, trientine, and sometimes oral zinc can prevent copper from accumulating. Copper content in drinking water should be checked, and people should be advised not to take any vitamin or mineral supplements containing copper.
Penicillamine is the most commonly used chelating drug but has considerable toxicity (eg, fever, rash, neutropenia, thrombocytopenia, proteinuria). Cross-reactivity may occur in people with penicillin allergy. Oral pyridoxine is given with penicillamine. Occasionally, use of penicillamine can cause worsening neurologic symptoms.
Appropriate dosing of penicillamine is based on measurement of urinary copper excretion and free serum copper levels.
Trientine hydrochloride, also a chelating drug, is an alternative treatment to penicillamine.
Zinc acetate can reduce intestinal copper absorption, thus preventing reaccumulation of copper in patients who cannot tolerate penicillamine or trientine or who have neurologic symptoms that do not respond to the other drugs. (CAUTION: Penicillamine or trientine must not be taken at the same time as zinc because either drug can bind zinc, forming a compound with no therapeutic effect.)
Poor long-term adherence to pharmacologic therapy is common. After 1 to 5 years of therapy, lower dose maintenance drug therapy can be considered. Regular follow-up care with an expert in liver disease is recommended.
Liver transplantation may be lifesaving for patients who have Wilson disease and fulminant hepatic failure or severe hepatic insufficiency refractory to drugs.
Treatment reference
1. Schilsky ML, Roberts EA, Bronstein JM, et al. A multidisciplinary approach to the diagnosis and management of Wilson disease: 2022 Practice Guidance on Wilson disease from the American Association for the Study of Liver Diseases. Hepatology. Published online December 7, 2022. doi:10.1002/hep.32801
Prognosis for Wilson Disease
Prognosis for patients with Wilson disease is usually good, unless disease is advanced before treatment begins.
Untreated Wilson disease is fatal, usually by age 30.
Key Points
Wilson disease is a rare, autosomal recessive disorder in which copper accumulates in various organs.
The disease manifests during childhood or adulthood, usually between ages 5 and 35.
Suspect the disorder in people with a family history of the disorder or unexplained hepatic, neurologic, or psychiatric abnormalities (including elevated transaminase levels).
Confirm the diagnosis primarily with a slit-lamp examination (for Kayser-Fleischer rings) and measurement of serum ceruloplasmin (which is low) and 24-hour urinary copper excretion (which is high).
Advise patients to follow a low-copper diet, and treat them with penicillamine, trientine, or, if these medications are intolerable or ineffective, oral zinc.
Drug Information for the Topic





