




La fibrosi cistica è una malattia ereditaria delle ghiandole esocrine che colpisce principalmente l'apparato gastrointestinale e respiratorio. Conduce a malattia polmonare cronica, insufficienza pancreatica esocrina, disturbi epatobiliari e livelli particolarmente elevati di elettroliti nel sudore. La diagnosi viene posta in caso di positività del test del sudore o dell'identificazione di 2 varianti geniche causanti la fibrosi cistica in pazienti con screening neonatale positivo o segni clinici caratteristici. Il trattamento è di supporto attraverso la cura multidisciplinare aggressiva con correttori a piccole molecole e potenziatori di targeting del difetto della proteina che regola la conduttanza della transmembrana della fibrosi cistica.
La fibrosi cistica è una malattia genetica potenzialmente mortale, che negli Stati Uniti si verifica in circa 1/3300 nascite di soggetti bianchi, 1/15 300 nascite di soggetti neri e 1/32 000 nascite di soggetti asiatici americani. Ci sono circa 40 000 persone con fibrosi cistica che vivono negli Stati Uniti, e circa 100 000 con diagnosi di fibrosi cistica nel mondo. Grazie al miglioramento delle cure e dell'aspettativa di vita, circa il 58% dei pazienti affetti da fibrosi cistica negli Stati Uniti è costituito da adulti (1).
Riferimento generale
1. Cystic Fibrosis Foundation Patient Registry 2021 Annual Data Report Bethesda, Maryland 2022 Cystic Fibrosis Foundation. Consultato il 20/10/2023.
Eziologia della fibrosi cistica
Circa il 3% della popolazione bianca è portatore sano della fibrosi cistica, che si trasmette con modalità autosomica recessiva. Il gene responsabile è stato localizzato sul braccio lungo del cromosoma 7. Codifica per una proteina associata alla membrana chiamata cystic fibrosis transmembrane conductance regulator (CFTR). La variante genetica più diffusa, F508del, si verifica in circa l'85% degli alleli della fibrosi cistica; sono state identificate > 2000 varianti del gene CFTR meno diffuse.
Il CFTR è un canale del cloro regolato dall'adenosina monofosfato ciclico, che regola il trasporto del cloro, del sodio e del bicarbonato attraverso le membrane epiteliali. Si ritengono probabili anche alcune funzioni aggiuntive. La malattia si manifesta solo negli omozigoti. Gli eterozigoti possono mostrare lievi anomalie nel trasporto elettrolitico, ma sono asintomatici.
Le varianti del CFTR sono state suddivise in 6 classi in base a come la variante influisca sulla funzione o sulla trasformazione della proteina CFTR. I pazienti con varianti di classe I, II o III sono considerati avere un più grave genotipo che si traduce in poca o nessuna funzione del CFTR, mentre i pazienti con 1 o 2 varianti di IV, V o VI classe sono considerati avere un genotipo più mite che si traduce in funzione del CFTR residua. Tuttavia, non c'è una stretta relazione tra varianti specifiche e le manifestazioni della malattia, e di conseguenza la valutazione clinica (ossia, della funzionalità degli organi) fornisce migliori indicazioni per la prognosi rispetto alla genotipizzazione. Le varianti del CFTR possono coinvolgere mutazioni frameshift (una delezione o inserimento in una sequenza di DNA che fa slittare il modo in cui una sequenza viene letta) o mutazioni nonsense (arresto-stop).
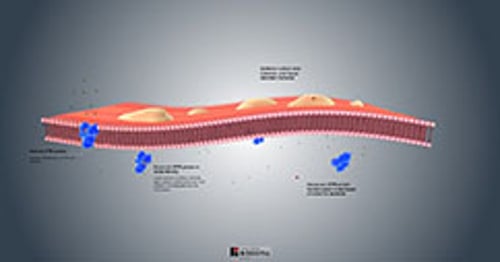
Fisiopatologia della fibrosi cistica
Quasi tutte le ghiandole esocrine sono interessate in modo più o meno grave. Le ghiandole possono
Essere progressivamente ostruite da muco vischioso presente nel lume (pancreas, ghiandole intestinali, dotti biliari intraepatici, colecisti e ghiandole sottomandibolari)
Alcune ghiandole appaiono istologicamente anomale e producono eccessive quantità di secrezioni (ghiandole tracheobronchiali e di Brunner)
Alcune ghiandole appaiono istologicamente normali, ma secernono eccessive quantità di sodio e cloro (ghiandole sudoripare, parotide, e piccole ghiandole salivari)
Apparato respiratorio
Sebbene i polmoni siano di solito istologicamente normali alla nascita, la maggior parte dei pazienti sviluppa segni di una malattia polmonare a partire dall'infanzia o dalla prima infanzia. L'ostruzione legata al muco e l'infezione batterica cronica, insieme a una risposta infiammatoria pronunciata, danneggiano le vie aeree, esitando in bronchiectasie e insufficienza respiratoria. Il decorso è caratterizzato da episodi di riacutizzazione infettive con progressivo deterioramento della funzionalità polmonare.
Il danno polmonare inizia probabilmente con un'ostruzione diffusa a carico delle vie aeree di piccolo calibro legata alle secrezioni mucose eccessivamente dense. La bronchiolite e i tappi di secrezioni mucopurulente a livello delle vie aeree sono secondari all'ostruzione e alle infezioni. Anche l'infiammazione cronica conseguente al rilascio di proteasi e citochine proinfiammatorie nelle vie aeree contribuisce al danno polmonare. Le alterazioni a carico delle vie aeree sono più frequenti rispetto a quelle a carico del parenchima, e l'enfisema non è marcato. Circa il 50% dei pazienti mostra un'iperreattività bronchiale che può rispondere ai broncodilatatori.
Nei pazienti con malattia polmonare avanzata, l'ipossiemia cronica determina ipertrofia muscolare delle arterie polmonari, ipertensione polmonare e ipertrofia ventricolare destra.
I polmoni della maggior parte dei pazienti sono colonizzati da batteri patogeni. Nelle prime fasi della malattia, il patogeno più diffuso è lo Staphylococcus aureus, ma con il progredire della malattia, il germe Pseudomonas aeruginosa, compresi i ceppi multiresistenti, viene frequentemente isolato. Un ceppo mucoide di P. aeruginosa è associato unicamente alla fibrosi cistica ed è legato a una prognosi peggiore rispetto ai Pseudomonas non mucoidi P. aeruginosa.
Negli Stati Uniti, la prevalenza di S. aureusmeticillino-resistente nelle vie aeree è ora circa del 25%; i pazienti con infezione da S. aureus meticillino-resistente (MRSA) cronica hanno un declino più rapido della funzionalità polmonare e tassi di sopravvivenza inferiori rispetto ai non infetti.
La colonizzazione con Burkholderia cepacia complex si verifica in circa il 2-3% dei pazienti e può essere associata a un più rapido deterioramento polmonare.
I micobatteri non tubercolari, tra cui il Mycobacterium avium complex e il M. abscessus, sono potenziali patogeni respiratori. La prevalenza è di circa il 14% e varia con l'età e la posizione geografica. Può essere difficile differenziare l'infezione dalla colonizzazione.
Altri patogeni respiratori comuni comprendono Stenotrophomonas maltophilia, Achromobacter xylosoxidans e Aspergillus spp.
Batteri anaerobi e virus respiratori comuni sono frequentemente presenti nel tratto respiratorio dei pazienti con fibrosi cistica, ma il loro ruolo nella progressione della malattia non è stato ben stabilito.
Gastrointestinale
Il pancreas, l'intestino e il sistema epatobiliare sono interessati frequentemente. La funzione pancreatica esocrina è compromessa nell'85-95% dei pazienti. Un'eccezione è rappresentata da un sottogruppo di pazienti che presentano alcune varianti del CFTR con funzione residua, in cui la funzione pancreatica è conservata. I pazienti con insufficienza pancreatica presentano malassorbimento di grassi, vitamine liposolubili e proteine. Il succo duodenale è patologicamente viscoso e mostra assenza o riduzione dell'attività enzimatica e ridotta concentrazione di bicarbonato; la tripsina e la chimotripsina sono assenti o ridotte nelle feci. La disfunzione pancreatica endocrina è meno comune, ma l'alterata tolleranza al glucosio o il diabete mellito sono presenti in circa il 2% dei bambini, nel 20% degli adolescenti, e fino al 50% degli adulti.
Il coinvolgimento dei dotti biliari con stasi e ostruzione biliare porta a fibrosi epatica asintomatica nel 30% dei pazienti. Circa il 3-4% dei pazienti progredisce verso la cirrosi biliare multinodulare irreversibile con varici e ipertensione portale, generalmente entro i 12 anni di età. L'insufficienza epatocellulare è un evento raro e tardivo. Vi è una maggiore incidenza di colelitiasi, che generalmente è asintomatica.
Le secrezioni intestinali viscose possono causare ileo da meconio nei neonati e talvolta ostruzione da meconio del colon. I bambini più grandi e gli adulti possono avere stipsi intermittente o cronica e occlusione intestinale.
Altri problemi gastrointestinali comprendono invaginazione, volvolo, prolasso rettale, ascesso periappendicolare, pancreatite, un aumentato rischio di cancro del tratto epatobiliare e del tratto digerente (compreso del pancreas), reflusso gastroesofageo, esofagite, e un'aumentata prevalenza di malattia di Crohn e celiachia.
Altro
L'infertilità si verifica nel 98% dei soggetti adulti di sesso maschile per l'alterato sviluppo dei dotti deferenti o per altre forme di azoospermia ostruttiva. Nelle donne, la fertilità è ridotta in qualche misura a causa della viscosità delle secrezioni cervicali, anche se molte donne riescono a portare a termine la gravidanza. L'esito della gravidanza sia per la madre che per il neonato è legato alla salute della madre.
Altre complicanze comprendono rinosinusite cronica, osteopenia/osteoporosi, depressione e ansia, dolore cronico, apnea ostruttiva del sonno, altri disturbi del sonno, calcoli renali, malattia renale cronica dialisi-dipendente (possibilmente collegata a trattamenti come verso la fibrosi cistica), anemia sideropenica, perdita dell'udito neurosensoriale e acufene causati dall'esposizione a farmaci ototossici (in particolare gli aminoglicosidi) e artralgia/artrite episodica.
Sintomatologia della fibrosi cistica
Apparato respiratorio
Il 50% dei pazienti non diagnosticato allo screening neonatale manifesta sintomi respiratori, spesso con esordio nell'infanzia. Sono comuni le infezioni ricorrenti o croniche che si manifestano con tosse, espettorazione e respiro sibilante. La tosse è il sintomo cronico più frequente, spesso accompagnata da produzione di espettorato, vomito e disturbi del sonno. Con il progredire della malattia, compaiono rientramenti intercostali, impiego dei muscoli respiratori accessori, torace a botte, ippocratismo digitale, cianosi e una tolleranza che diminuisce per l'esercizio. L'interessamento delle vie respiratorie superiori comprende la poliposi nasale e la rinosinusite cronica o ricorrente.
Le complicanze polmonari comprendono pneumotorace, infezione da micobatteri non tubercolari, emottisi, aspergillosi broncopolmonare allergica e insufficienza cardiaca destra secondaria ad ipertensione polmonare.
Gastrointestinale
L'ileo da meconio, causato dall'ostruzione intestinale da parte di meconio viscoso, è la manifestazione più precoce ed è presente in circa il 10-20% dei neonati affetti da fibrosi cistica. Si manifesta tipicamente con distensione addominale, vomito e incapacità di evacuare il meconio. Alcuni neonati presentano perforazione intestinale, con segni di peritonite e shock. I neonati con sindrome da tappo di meconio presentano una ritardata evacuazione del meconio. Possono manifestare segni simili a quelli dell'occlusione o sintomi molto lievi e transitori che passano inosservati. I pazienti anziani possono andare incontro ad episodi di stipsi o sviluppare episodi ricorrenti e a volte cronici di occlusione parziale o completa dell'intestino tenue o crasso (sindrome da ostruzione dell'intestino distale). I sintomi includono dolore addominale crampiforme, cambiamenti nelle modalità di evacuazione delle feci, diminuzione dell'appetito, e talvolta vomito.
Nei neonati senza ileo da meconio, l'esordio della malattia può manifestarsi con un tardivo recupero del peso alla nascita e un aumento ponderale inadeguato a 4-6 settimane di vita.
Occasionalmente, i neonati che sono denutriti, soprattutto se allattati con formule ipoallergeniche o di soia, presentano edema generalizzato secondario al malassorbimento proteico.
L'insufficienza pancreatica diventa clinicamente evidente nell'infanzia e può essere progressiva. Le manifestazioni includono frequente evacuazione di abbondanti feci oleose e maleodoranti; addome prominente; scarso accrescimento con riduzione del tessuto sottocutaneo e della massa muscolare nonostante un appetito normale o vorace. Alcune manifestazioni cliniche possono verificarsi a causa della carenza di vitamine liposolubili.
Il prolasso rettale può verificarsi nei lattanti e nei bambini non trattati. Il reflusso gastroesofageo è relativamente comune tra bambini e adulti.
Altro
L'eccessiva sudorazione durante un periodo caldo o in corso di febbre può causare episodi di disidratazione iponatriemica/ipocloremica e insufficienza circolatoria. Nelle zone con clima arido i lattanti possono andare incontro a un'alcalosi metabolica cronica. La formazione di cristalli di sale e il sapore salato sulla pelle sono molto che suggeriscono fibrosi cistica.
Gli adolescenti possono manifestare ritardo di crescita e pubertà ritardata.
Diagnosi della fibrosi cistica
Screening neonatale
La fibrosi cistica può essere sospettata per lo screening prenatale positivo, per l'anamnesi familiare o per le manifestazioni cliniche
È confermata da un test del sudore che mostri elevate concentrazioni di cloro in 2 occasioni
L'identificazione di 2 varianti che causano la fibrosi cistica (1 su ciascun cromosoma) è compatibile con la diagnosi
Raramente può essere confermata, in casi atipici, dimostrando l'anomalo trasporto degli ioni attraverso l'epitelio nasale o le misurazioni anomale della corrente intestinale
La maggior parte dei casi di fibrosi cistica viene identificata con lo screening neonatale, ma fino al 10% non giunge alla diagnosi fino all'adolescenza o alla giovane età adulta. Nonostante i progressi nella diagnostica genetica, il test del sudore rimane l'esame più accurato per la conferma della diagnosi di fibrosi cistica nella maggior parte dei casi in quanto sensibile, specifico, semplice e disponibile.
Screening neonatale
Lo screening neonatale universale per la fibrosi cistica è ormai standard negli Stati Uniti. Lo screening si basa sulla rilevazione di un'elevata concentrazione di tripsinogeno immunoreattivo nel sangue.
Esistono 2 metodi di follow up in caso di riscontro di elevati livelli di tripsinogeno immunoreattivo. In un metodo, si esegue un secondo test per il tripsinogeno immunoreattivo, che, se elevato, è seguito da un test del sudore. Nell'altro metodo, utilizzato più comunemente, un livello elevato di tripsinogeno immunoreattivo è seguito dalla ricerca delle mutazioni del CFTR e, in caso vengano identificate 1 o 2 varianti, si effettua anche il test del sudore. Per la diagnosi, entrambi i metodi hanno una sensibilità del 90-95%.
Test del sudore
In questo test, viene stimolata localmente la sudorazione con la pilocarpina, la quantità di sudore viene misurata e viene quindi analizzata la sua concentrazione di cloro. Sebbene la concentrazione di cloro nel sudore aumenti lievemente con l'età, il test del sudore è valido a qualsiasi età:
Normale: ≤ 30 mEq/L (≤ 30 mmol/L) (la fibrosi cistica è improbabile.)
Intermedio: da 30 a 59 mEq/L (da 30 a 59 mmol/L) (la fibrosi cistica è possibile.)
Anormale: ≥ 60 mEq/L (≥ 60 mmol/L) (Questo risultato è coerente con la fibrosi cistica.)
I risultati sono già attendibili dopo le prime 48 h di vita, ma prima delle 2 settimane di vita può essere difficile ottenere un adeguato campione di sudore (> 75 mg su carta bibula o > 15 mcL con capillare). Raramente si possono ottenere risultati falsamente negativi, che sono dovuti alla presenza di edema e ipoproteinemia o inadeguata quantità di sudore. I risultati falsamente positivi sono dovuti, in genere, ad errori tecnici. Un aumento transitorio della concentrazione di cloro nel sudore può derivare da situazioni di deprivazione psicosociale (p. es., maltrattamento, trascuratezza) e può verificarsi in pazienti affetti da anoressia nervosa. Un risultato positivo del test del sudore deve essere confermato da un secondo test del sudore o dall'identificazione di 2 varianti causanti fibrosi cistica.
Risultati borderline del test del sudore
Un piccolo sottogruppo di pazienti presenta un fenotipo lieve o parziale di fibrosi cistica e valori di CI nel sudore che sono costantemente borderline o normali. Inoltre, esistono pazienti che hanno manifestazioni a carico di singoli organi come pancreatite cronica o ricorrente, bronchiectasie isolate o assenza congenita bilaterale dei vasi deferenti insieme a segni che suggeriscono funzione anomala del CFTR. Non soddisfano i criteri per la diagnosi di fibrosi cistica e sono classificati come affetti da un disturbo correlato al CFTR. In alcuni di questi pazienti, la diagnosi di fibrosi cistica può essere confermata dall'identificazione di 2 varianti causanti fibrosi cistica, una in ciascun cromosoma. Se non vengono identificate 2 varianti causa di fibrosi cistica, possono essere utili ulteriori indagini tra le quali lo studio per immagini e funzionale del pancreas, la TC del torace ad alta risoluzione, la TC dei seni paranasali, le prove di funzionalità respiratoria, la valutazione urogenitale nei maschi, e il lavaggio broncoalveolare con valutazione della flora microbica.
Ulteriori indagini diagnostiche potenzialmente utili comprendono il sequenziamento completo del gene CFTR e la misurazione della differenza di potenziale transepiteliale nasale (basata sull'osservazione di un maggiore riassorbimento di sodio attraverso l'epitelio, che è relativamente impermeabile al cloro nei pazienti con fibrosi cistica) e la misurazione delle correnti intestinali.
Sindrome metabolica correlata al CFTR e diagnosi positiva per fibrosi cistica, non definitiva
I neonati che hanno un risultato di screening neonatale positivo e l'evidenza di una possibile disfunzione del CFTR, ma che non soddisfano i criteri diagnostici per la fibrosi cistica sono classificati come affetti da sindrome metabolica correlata al CFTR (CFTR-related metabolic syndrome [CRMS] anche chiamati CF screen positive, inconclusive diagnosis [CFSPID (screening della fibrosi cistica positiva, diagnosi inconcludente)]). La CRMS/CFSPID, viene diagnosticata nei neonati che hanno un risultato di screening neonatale positivo, sono asintomatici e presentano qualsiasi dei seguenti segni:
Concentrazioni di cloruro nel sudore nella gamma intermedia e 0 o 1 variante che provoca fibrosi cistica
Concentrazioni di cloruro nel sudore nel range di normalità e 2 varianti del CFTR, almeno una delle quali con conseguenze fenotipiche non chiare
La maggior parte dei bambini con CRMS/CFSPID rimane in buona salute, ma nel tempo intorno al 10% svilupperà sintomi e soddisferà i criteri per la diagnosi di fibrosi cistica o una patologia correlata alla fibrosi cistica. I pazienti con CRMS/CFSPID devono essere valutati e monitoratiregolarmente in un centro di cura per la fibrosi cistica.
Studio del pancreas
Al momento della diagnosi deve essere valutata la funzionalità pancreatica, generalmente attraverso la misurazione della concentrazione dell'elastasi pancreatica umana nelle feci. La misurazione dell'elastasi pancreatica umana è valida anche in presenza di enzimi pancreatici esogeni. I lattanti che inizialmente hanno una funzionalità pancreatica adeguata, ma presentano 2 varianti "gravi", devono essere valutati periodicamente per rilevare la progressione verso l'insufficienza pancreatica.
Valutazione delle vie respiratorie
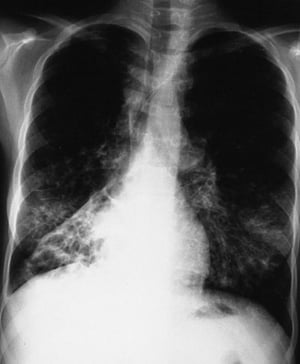
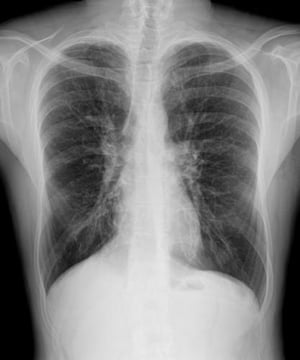
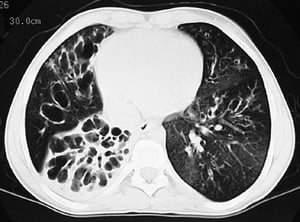
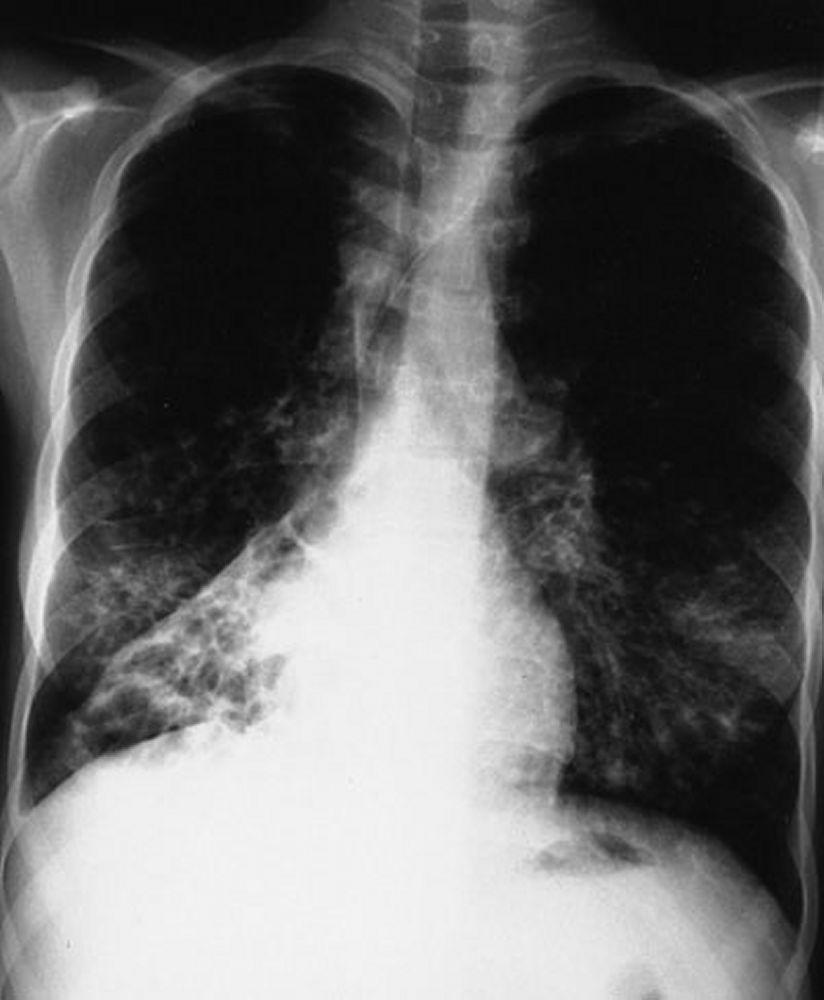
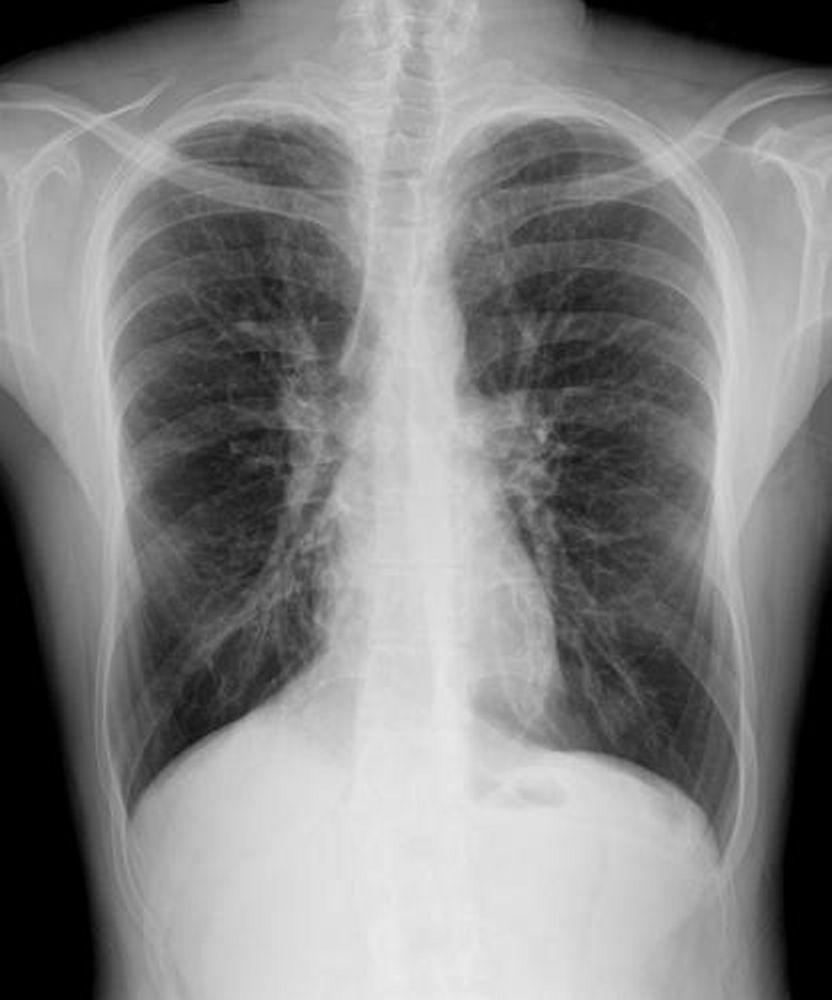
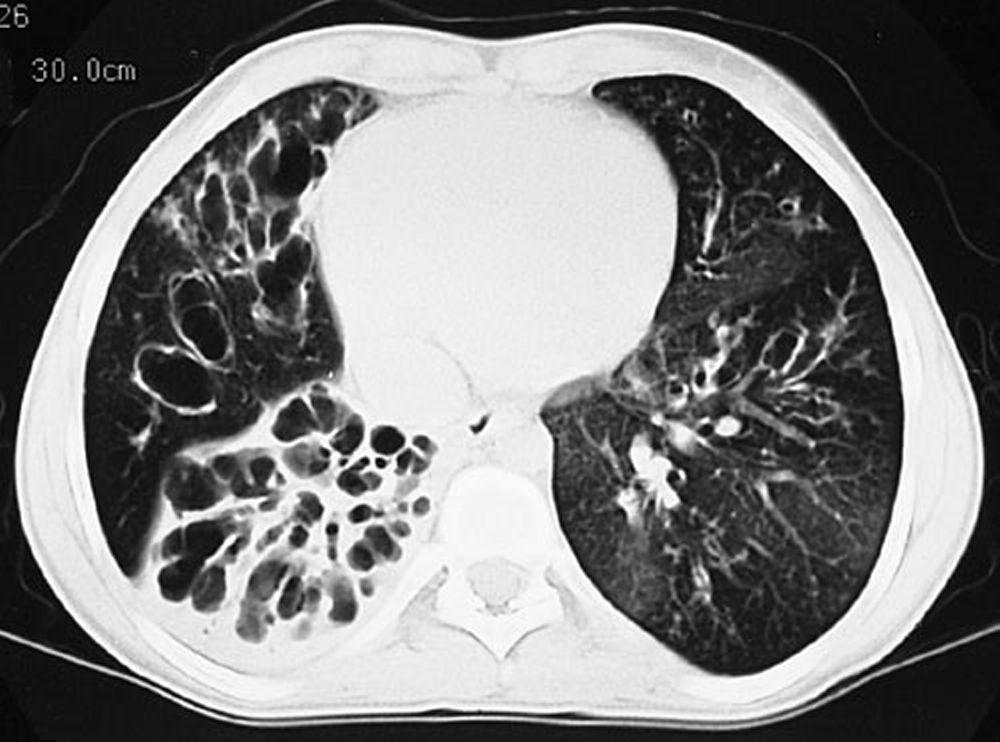
L'imaging del torace viene eseguita in caso di peggioramento della funzionalità polmonare o di riacutizzazione e, di routine, ogni 1 o 2 anni. La TC ad alta risoluzione del torace può essere utile per definire con maggiore precisione l'estensione del danno polmonare e per rilevare le alterazioni meno evidenti delle vie aeree più periferiche. La RX e la TC del torace possono mettere in evidenza iperespansione, addensamento mucoide e ispessimento della parete bronchiale come reperti più precoci. Successivamente saranno visibili aree di infiltrazione, atelettasia e adenopatia ilare. Con il progredire della malattia si verificano atelettasie segmentarie o lobari, formazione di cisti, bronchiectasie, ipertrofia dell'arteria polmonare e del ventricolo destro. Sono caratteristiche le opacità ramificate, digitiformi, che rappresentano la dilatazione dei bronchi ostruiti da secrezioni mucoidi.
Lo studio TC dei seni paranasali è indicato in pazienti con sintomatologia sinusale significativa o polipi nasali nei quali viene presa in considerazione la chirurgia endoscopica. Queste indagini evidenziano quasi sempre un'opacizzazione persistente dei seni paranasali.
By permission of the publisher. Da Berman L: Atlas of Anesthesia: Critical Care. Edited by R Miller (series editor) and RR Kirby. Philadelphia, Current Medicine, 1997.
PHOTOSTOCK-ISRAEL/SCIENCE PHOTO LIBRARY
By permission of the publisher. Da Berman L: Atlas of Anesthesia: Critical Care. Edited by R Miller (series editor) and RR Kirby. Philadelphia, Current Medicine, 1997.

By permission of the publisher. Da Berman L: Atlas of Anesthesia: Critical Care. Edited by R Miller (series editor) and RR Kirby. Philadelphia, Current Medicine, 1997.

PHOTOSTOCK-ISRAEL/SCIENCE PHOTO LIBRARY

By permission of the publisher. Da Berman L: Atlas of Anesthesia: Critical Care. Edited by R Miller (series editor) and RR Kirby. Philadelphia, Current Medicine, 1997.
Le prove di funzionalità respiratoria sono i migliori indicatori delle condizioni cliniche e della risposta alla terapia dei pazienti. In pazienti sopra i 5 anni, la spirometria deve essere eseguita regolarmente e in caso di peggioramento clinico. Nei neonati, lo stato respiratorio può essere monitorato utilizzando una tecnica di compressione toracoaddominale a volume elevato, che genera una curva flusso-volume parziale. Nei bambini da 3 a 6 anni di età, la procedura di washout con respiro multiplo può essere utilizzata per generare un indice di clearance polmonare come misura della disomogeneità della ventilazione (1).
Le prove di funzionalità respiratoria fatte tramite spirometria indicano
Una riduzione della capacità vitale forzata (CVF), del volume espiratorio forzato in 1 secondo (FEV1), del flusso espiratorio forzato tra il 25% e il 75% del volume espirato (FEF25-75), e del rapporto FEV1/CVF
Un aumento del volume residuo e il rapporto tra volume residuo e capacità polmonare totale
Il 50% dei pazienti presenta un'ostruzione reversibile delle vie aeree, come evidenziato dal miglioramento dei parametri della funzionalità respiratoria dopo la somministrazione di un broncodilatatore per via inalatoria.
Le colture dell'espettorato e lo screening orofaringei devono essere eseguiti almeno 4 volte/anno, specialmente nei pazienti non ancora colonizzati con P. aeruginosa. La broncoscopia e il lavaggio broncoalveolare sono indicati quando è importante definire con precisione la flora microbica delle vie aeree inferiori del paziente (p. es., per mirare la scelta degli antibiotici) o per rimuovere tappi di muco denso.
Screening del portatore
Lo screening del portatore sano di fibrosi cistica è disponibile negli Stati Uniti ed è consigliato per le coppie che stanno pianificando una gravidanza o effettuando accertamenti prenatali. Se entrambi i potenziali genitori risultano portatori di una variante del CFTR, si può effettuare un'analisi prenatale del feto mediante prelievo di villi coriali o amniocentesi. La consulenza prenatale in questi casi è complicata dall'ampia variabilità fenotipica della fibrosi cistica e dalle informazioni incomplete sulle conseguenze cliniche di molte delle varianti del gene CFTR che vengono identificate attraverso lo screening.
Riferimento relativo alla diagnosi
1. Stanojevic S, Davis SD, Retsch-Bogart G, et al: Progression of lung disease in preschool patients with cystic fibrosis. Am J Respir Crit Care Med 195:1216–1225, 2017. doi: 10.1164/rccm.201610-2158OC
Trattamento della fibrosi cistica
Supporto omnicomprensivo e multidisciplinare
Antibiotici, farmaci inalatori per le secrezioni delle piccole vie aeree e manovre fisiche per eliminare le secrezioni delle vie aeree
Broncodilatatori per via inalatoria e talvolta corticosteroidi per i pazienti che rispondono al trattamento
Di solito supplementazione con enzimi pancreatici e vitamine
Dieta ad alto contenuto calorico (che a volte richiede alimentazione enterale supplementare)
Nei pazienti con varianti specifiche, modulatori del regolatore della conduttanza transmembrana della fibrosi cistica (CFTR modulators) consistono in un potenziatore del CFTR o in una combinazione di correttori del CFTR e un potenziatore del CFTR
È essenziale un programma terapeutico globale e intensivo, guidato da personale medico con particolare esperienza nel campo, che operi in un team multidisciplinare che comprenda medici, infermieri, nutrizionisti, fisioterapisti e riabilitatori, professionisti della salute mentale, farmacisti e assistenti sociali. Lo scopo della terapia è mantenere un adeguato stato nutrizionale, prevenire o trattare in modo aggressivo le complicanze polmonari o altre complicanze, incoraggiare l'attività fisica e provvedere a un sostegno psicosociale adeguato. Il regime di trattamento è complesso e può richiedere fino a 2 h ogni giorno. Con un supporto appropriato, la maggior parte dei pazienti può trovare un buon inserimento a scuola e in famiglia.
(Vedi anche the Cystic Fibrosis Foundation's comprehensive treatment guidelines for all age groups.)
Trattamento delle manifestazioni respiratorie
Il trattamento delle manifestazioni respiratorie è focalizzato sulla prevenzione dell'ostruzione delle vie aeree, sulla profilassi e sul controllo delle infezioni respiratorie. La profilassi contro le infezioni polmonari comprende il mantenimento dell'immunità da pertosse, Haemophilus influenzae, varicella, Streptococcus pneumoniae, e morbillo; la vaccinazione antinfluenzale annuale; e la vaccinazione contro il COVID-19 secondo le current recommendations dell'Advisory Committee on Immunization Practices (ACIP). Nei pazienti esposti al virus dell'influenza, può essere utilizzato un inibitore della neuraminidasi a scopo profilattico o ai primi segni di infezione. La somministrazione del nirsevimab, o quando non disponibile, del palivizumab in lattanti con fibrosi cistica per la prevenzione dell'infezione da virus respiratorio sinciziale si è dimostrata sicura, ma la sua efficacia non è stata documentata.
Si raccomanda una terapia a lungo termine con inalazione giornaliera di dornasi alfa (deossiribonucleasi umana ricombinante) o con soluzione salina ipertonica al 7% (1, 2), che ha dimostrato rallentare il tasso di declino della funzione polmonare e diminuire la frequenza delle riacutizzazioni del tratto respiratorio (3).
Al momento della diagnosi vengono raccomandate le manovre per la clearance delle vie aeree basate su drenaggio posturale, percussioni, vibrazioni e tosse assistita (fisioterapia toracica), e devono essere eseguite regolarmente, con costanza. Nei pazienti anziani, possono essere efficaci altre tecniche di drenaggio delle vie aeree, come cicli di respirazione attiva, drenaggio autogeno, dispositivi con pressione espiratoria positiva e terapia con il sistema Vest (oscillazione ad alta frequenza della parete del torace). Si raccomanda un regolare un esercizio fisico aerobico, che può anche contribuire alla fisioterapia. Per i pazienti con apnea ostruttiva del sonno, la ventilazione continua a pressione positiva può essere utile.
Per i pazienti che presentano ostruzione reversibile delle vie aeree, i broncodilatatori possono essere somministrati per via inalatoria. Di solito, i corticosteroidi somministrati per via inalatoria non risultano efficaci. L'ossigenoterapia è indicata per i pazienti con grave insufficienza respiratoria e ipossiemia.
Tipicamente, la ventilazione meccanica o l'ossigenazione extracorporea a membrana non sono indicate per i pazienti con insufficienza respiratoria cronica. Il loro uso è generalmente limitato ai pazienti in buone condizioni che presentano complicanze respiratorie reversibili, in associazione alla chirurgia toracica, o nei pazienti in cui è imminente il trapianto polmonare. Può essere utile anche la ventilazione non invasiva a pressione positiva con maschera nasale o facciale.
Gli espettoranti per via orale vengono talvolta usati, ma pochi dati ne supportano l'efficacia. I sedativi della tosse devono essere sconsigliati.
Lo pneumotorace può essere trattato con il drenaggio toracostomico. Nello pneumotorace ricorrente, i trattamenti più efficaci sono la toracotomia a cielo aperto o la toracoscopia con resezione delle bolle pleuriche e abrasione meccanica della superficie pleurica.
L'emottisi da lieve a moderata viene trattata con antibiotici (per via orale/aerosol o EV a seconda della gravità dell'emottisi e della gravità dell'infezione) e la clearance delle vie aeree. L'emottisi massiva o ricorrente viene trattata con l'embolizzazione delle arterie bronchiali o raramente con la resezione polmonare focale.
I corticosteroidi per via orale sono indicati nei lattanti con forme protratte di bronchiolite e nei pazienti con broncospasmo refrattario, aspergillosi broncopolmonare allergica, e complicanze infiammatorie (p. es., artrite e vasculite). L'impiego a lungo termine della terapia corticosteroidea a giorni alterni può rallentare il declino della funzionalità respiratoria, tuttavia, a causa delle complicanze legate agli steroidi, essa non è raccomandata come trattamento routinario. I pazienti che ricevono corticosteroidi devono essere monitorati attentamente per l'eventuale comparsa di diabete e rallentamento dell'accrescimento.
L'aspergillosi broncopolmonare allergica viene trattata anche con corticosteroidi sistemici e con un farmaco antimicotico orale.
È stato dimostrato che l'ibuprofene, somministrato per alcuni anni in dosi sufficienti a raggiungere una concentrazione di picco plasmatico compresa tra 50 e 100 mcg/mL (242,4 e 484,8 micromol/L), riduce la velocità del declino della funzionalità respiratoria, soprattutto nei bambini dai 5 ai 13 anni. La dose appropriata deve essere individualizzata sulla base di studi di farmacocinetica.
La rinosinusite cronica è molto frequente. Le opzioni terapeutiche comprendono irrigazione con soluzione fisiologica nasale, irrigazione nasale isotonica a bassa pressione, nebulizzazione intranasale della dornase alfa e antibiotici topici senonasali. La chirurgia sinusale può essere utile nei casi refrattari alla gestione medica. Si raccomanda uno spray di corticosteroidi intranasale per trattare la rinite allergica.
Modulatori della conduttanza transmembrana della fibrosi cistica
I farmaci per la correzione ed il potenziamento del CFTR, sono indicati per circa il 90% delle varianti che colpiscono i pazienti con fibrosi cistica. I modulatori del gene CFTR non sono disponibili per i pazienti in classe I e con mutazioni frameshift (inserzioni o delezioni) e nonsense.
L'ivacaftor è un farmaco orale a piccole molecole somministrato cronicamente che potenzia il canale ionico del CFTR difettoso in pazienti con specifiche varianti dello stesso. Può essere usato in pazienti di 1 mese e più grandi portatori di almeno 1 copia di una specifica variante potenziata dall'ivacaftor.
Il lumacaftor, il tezacaftor e l'elexacaftor sono farmaci orali a piccole molecole che correggono in parte la proteina CFTR difettosa alterando il ripiegamento della proteina in pazienti che portano la variante F508del o altre varianti specifiche.
La combinazione di lumacaftor e ivacaftor può essere somministrata a pazienti di 1 anno e più grandi portatori di 2 copie della variante F508del.
La combinazione di tezacaftor e ivacaftor può essere somministrata a pazienti di 6 anni e più grandi portatrici di 2 copie della variante F508del o di altre varianti specifiche.
La tripla combinazione di elexacaftor, tezacaftor e ivacaftor può essere somministrata a pazienti di 2 anni e più grandi portatori di almeno 1 copia della variante F508del o 1 copia di certe varianti rare (4, 5).
Questi farmaci possono migliorare la funzionalità respiratoria, aumentare il peso, migliorare la funzione pancreatica esocrina, diminuire la frequenza delle esacerbazioni polmonari e delle ospedalizzazioni, migliorare la qualità della vita ridurre e talvolta normalizzare le concentrazioni di cloro nel sudore (6). Le indicazioni per ivacaftor, lumacaftor/ivacaftor, tezacaftor/ivacaftor ed elexacaftor/tezacaftor/ivacaftor sono basate sulle varianti del gene CFTR del paziente e sull'età e stanno cambiando rapidamente. Sebbene tutti questi farmaci possano essere utili, solo l'ivacaftor e la combinazione di elexacaftor, tezacaftor ed ivacaftor sono considerati come una terapia modulatoria altamente efficace.
Trattamento e prevenzione delle infezioni
Per le esacerbazioni polmonari lievi, deve essere prescritto un breve ciclo di antibiotici orali sulla base dei test colturali e di sensibilità. Per lo stafilococco meticillino-sensibile, i farmaci di scelta sono una penicillina penicillinasi-resistente (p. es., dicloxacillina) e una cefalosporina (p. es., cefalexina) o il trimetoprim/sulfametossazolo. Possono essere impiegati anche eritromicina, amoxicillina/clavulanato, una tetraciclina o linezolid. Per i pazienti colonizzati dallo S. aureus meticillino-resistente (MRSA), può essere efficace un ciclo orale di trimetoprim/sulfametossazolo, clindamicina, linezolid o tetraciclina. Per i pazienti colonizzati con P. aeruginosa, un breve ciclo di tobramicina o aztreonam per via inalatoria (p. es., 4 settimane) e/o un fluorochinolone per via orale (p. es., da 2 a 3 settimane) possono essere efficaci. I fluorochinolonici sono stati utilizzati in modo sicuro nei bambini piccoli.
Per le esacerbazioni polmonari da moderate a gravi, specialmente nei pazienti colonizzati da P. aeruginosa, è consigliata la terapia antibiotica EV. I pazienti spesso richiedono il ricovero in ospedale, ma casi attentamente selezionati possono tranquillamente ricevere parte della terapia a domicilio. Si somministra per EV l'associazione dell'aminoglicoside tobramicina (o a volte l'amikacina) più cefalosporina, penicillina ad ampio spettro, fluorochinolone, o monobactam con attività antipseudomonas, in genere per 2 settimane. Dosi più elevate possono essere necessarie per raggiungere concentrazioni sieriche accettabili. Per l'aumentata clearance renale nei pazienti affetti da fibrosi cistica, possono essere necessarie dosi elevate di alcune penicilline per ottenere dei livelli ematici adeguati. Per i pazienti colonizzati con S. aureus meticillino-resistente (MRSA), la vancomicina o il linezolid possono essere aggiunti al regime EV.
L'eradicazione dell'infezione cronica da P. aeruginosa è difficile. È stato dimostrato, comunque, che l'antibioticoterapia precoce nel periodo in cui le vie aeree cominciano a essere infettate da parte di P. aeruginosa può essere efficace nell'eradicare il microrganismo per alcuni periodi. Nei pazienti colonizzati cronicamente da P. aeruginosa, la terapia antibiotica per via inalatoria è in grado di migliorare i parametri clinici e, se possibile, di ridurre la colonizzazione batterica nelle vie aeree (7). L'impiego a lungo termine, a mesi alterni, della tobramicina o dell'aztreonam per via inalatoria insieme alla terapia continuativa per 3 volte/settimana (ogni mese) con l'azitromicina per via orale può risultare efficace nel migliorare o stabilizzare la funzionalità respiratoria e nel ridurre la frequenza delle esacerbazioni polmonari.
I pazienti con infezione clinicamente significativa da parte di micobatteri non tubercolari possono richiedere una terapia a lungo termine con una combinazione di antibiotici per via orale, per via inalatoria e per EV.
I pazienti con aspergillosi broncopolmonare allergica o infezione aspergillare delle basse vie aeree possono richiedere una terapia orale o EV prolungata con un antifungino azolico e/o corticosteroidi sistemici.
Trattamento delle manifestazioni gastrointestinali
L'occlusione intestinale neonatale può essere talvolta risolta con un clisma con mezzo di contrasto radiopaco iperosmolare o isosmolare; in altrimenti, può essere necessaria l'enterostomia chirurgica per drenare il meconio viscoso dal lume intestinale. Dopo il periodo neonatale, gli episodi di occlusione intestinale parziale (sindrome da occlusione intestinale distale) possono essere trattati con clisma con mezzo di contrasto radiopaco iperosmolare o isosmolare o con acetilcisteina oppure mediante la somministrazione per via orale di una soluzione per il lavaggio intestinale. Per prevenire tali episodi può essere utile la somministrazione di emollienti delle feci come il sulfosuccinato dioctil-sodico (docusato) o il lattulosio.
L'acido ursodesossicolico, un acido biliare idrofilo, viene spesso utilizzato in pazienti con malattia epatica causata dalla fibrosi cistica, ma poche evidenze ne supportano l'efficacia per la prevenzione della progressione dalla stasi biliare alla cirrosi.
La terapia enzimatica pancreatica sostitutiva deve essere somministrata con l'assunzione dei pasti e degli spuntini nei pazienti con insufficienza pancreatica. Le preparazioni enzimatiche più efficaci contengono pancreolipasi in microsferule o micropastiglie gastroresistenti, sensibili al pH. Per i lattanti le capsule vengono aperte e il loro contenuto viene mescolato con il cibo acido. Dopo l'infanzia, viene utilizzato un dosaggio basato sul peso. Si devono evitare dosi di lipasi > 2500 UI/kg/pasto o > 10 000 UI/kg/die perché dosi elevate di enzimi sono associate con la colonpatia fibrosante. Nei pazienti con necessità di elevati dosaggi di enzimi, la loro efficacia può essere migliorata con la soppressione della secrezione acida mediante l'uso di un anti-H2 o di un inibitore della pompa protonica.
La terapia dietetica comporta l'assunzione di calorie e proteine sufficienti a garantire una crescita normale; può essere necessario dal 30 al 50% in più del fabbisogno dietetico giornaliero raccomandato (vedi tabella Apporto dietetico di riferimento consigliato per alcuni macronutrienti). La terapia alimentare comprende anche un introito totale di grassi da normale ad elevato per aumentare il contenuto calorico della dieta, un supplemento multivitaminico idrosolubile in quantità doppia rispetto ai fabbisogni giornalieri raccomandati, una supplementazione di vitamina D3 (colecalciferolo) nei pazienti con carenza o insufficienza da vitamina D, e una supplementazione di sale durante l'infanzia e nei periodi di stress termico e di incremento della sudorazione. I lattanti sottoposti ad antibioticoterapia ad ampio spettro e i pazienti con interessamento epatico ed emottisi devono ricevere un supplemento aggiuntivo di vitamina K. Nei lattanti con grave malassorbimento, al posto del latte vaccino intero modificato devono essere utilizzate formule costituite da idrolisati proteici e con acidi grassi a catena media. Per aumentare l'apporto calorico si possono somministrare polimeri del glucosio e acidi grassi a catena media.
Nei pazienti che non riescono a mantenere uno stato nutrizionale adeguato, l'integrazione enterale mediante gastrostomia o digiunostomia può migliorare un normale accrescimento e stabilizzare la funzione respiratoria (vedi Panoramica sul supporto nutrizionale). L'uso di stimolanti dell'appetito per promuovere la crescita può essere utile in alcuni pazienti.
Trattamento delle altre manifestazioni
Il diabete correlato alla fibrosi cistica è causato da deficit di insulina e condivide le caratteristiche del diabete di tipo 1 e 2. L'unico trattamento raccomandato è l'insulina. La gestione comprende un regime di terapia con somministrazione di insulina, consulenza nutrizionale, un programma di formazione di auto-gestione del diabete, e il monitoraggio per le complicanze microvascolari. Il piano terapeutico deve essere realizzato in collaborazione con un endocrinologo e un dietista con esperienza nel trattamento sia della fibrosi cistica che del diabete.
I pazienti con insufficienza cardiaca destra sintomatica devono essere sottoposti a terapia con diuretici, riduzione di sale nella dieta e ossigeno.
L'ormone della crescita ricombinante umano (rhGH) può migliorare la funzionalità respiratoria, aumentare l'altezza e il peso e il contenuto minerale osseo, e ridurre il tasso di ospedalizzazione. Tuttavia, l'rhGH non viene comunemente utilizzato a causa dei costi e dei disagi aggiuntivi.
La chirurgia può essere indicata per le bronchiectasie localizzate o le atelettasie che non possono essere efficacemente trattate con farmaci, per la poliposi nasale, per la rinosinusite cronica, per il sanguinamento dalle varici esofagee secondarie all'ipertensione portale, per la colecistopatia, per l'occlusione intestinale dovuta a volvolo o invaginazione che non regredisce con terapia medica.
Il trapianto di fegato è stato eseguito con successo in pazienti con epatopatia allo stadio terminale.
Spesso è necessaria una discussione riguardo al trapianto di polmone. Nel considerare il trapianto, i pazienti hanno bisogno di confrontare i vantaggi di una maggiore sopravvivenza grazie al trapianto con l'incertezza di ricevere il trapianto stesso e il peso continuo (ma diverso) di vivere con un trapianto d'organo. Il trapianto polmonare bilaterale da cadavere e il trapianto lobare da donatore vivo sono stati eseguiti con successo in pazienti con malattia polmonare avanzata. Il trapianto combinato di fegato e polmone è stato eseguito in pazienti con epatopatia e malattia polmonare allo stadio terminale.
Il trapianto di polmone bilaterale per la malattia polmonare grave sta diventando sempre più di routine e di maggior successo con l'aumentare dell'esperienza e il miglioramento delle tecniche. Tra gli adulti con fibrosi cistica, la sopravvivenza mediana post-trapianto è di circa 9 anni.
Riferimenti relativi al trattamento
1. Flume PA, O'Sullivan BP, Robinson KA, et al. Cystic fibrosis pulmonary guidelines: chronic medications for maintenance of lung health. Am J Respir Crit Care Med 2007;176(10):957-969. doi:10.1164/rccm.200705-664OC
2. Mogayzel PJ Jr, Naureckas ET, Robinson KA, et al. Cystic fibrosis pulmonary guidelines. Chronic medications for maintenance of lung health. Am J Respir Crit Care Med 2013;187(7):680-689. doi:10.1164/rccm.201207-1160oe
3. Stahl M, Wielpütz MO, Ricklefs I, et al. Preventive Inhalation of Hypertonic Saline in Infants with Cystic Fibrosis (PRESIS). A Randomized, Double-Blind, Controlled Study. Am J Respir Crit Care Med 2019;199(10):1238-1248. doi:10.1164/rccm.201807-1203OC
4. Heijerman HGM, McKone EF, Downey DG, et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial [published correction appears in Lancet 2020 May 30;395(10238):1694]. Lancet 2019;394(10212):1940-1948. doi:10.1016/S0140-6736(19)32597-8
5. Middleton PG, Mall MA, Dřevínek P, et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N Engl J Med 2019;381(19):1809-1819. doi:10.1056/NEJMoa1908639
6. Taylor-Cousar JL, Robinson PD, Shteinberg M, Downey DG. CFTR modulator therapy: transforming the landscape of clinical care in cystic fibrosis. Lancet 2023;402(10408):1171-1184. doi:10.1016/S0140-6736(23)01609-4
7. Mogayzel PJ Jr, Naureckas ET, Robinson KA, et al. Cystic Fibrosis Foundation pulmonary guideline. pharmacologic approaches to prevention and eradication of initial Pseudomonas aeruginosa infection. Ann Am Thorac Soc 2014;11(10):1640-1650. doi:10.1513/AnnalsATS.201404-166OC
Prognosi della fibrosi cistica
Il decorso è estremamente influenzato dal grado di interessamento polmonare. Il deterioramento della funzione polmonare nel tempo, generalmente caratterizzato da bronchiectasie progressive, porta alla debilitazione e, infine, aumenta il rischio di morte, di solito a causa di una combinazione di insufficienza respiratoria e cuore polmonare.
La prognosi è migliorata progressivamente negli ultimi 5 anni, soprattutto per l'introduzione di una diagnosi precoce e di una terapia aggressiva prima dell'insorgenza di danni polmonari irreversibili. L'età mediana al decesso nel 2021 era di 33,9 anni. La sopravvivenza mediana prevista negli Stati Uniti per i bambini nati nel 2021 è di 65,6 anni. La sopravvivenza è significativamente più lunga nei pazienti non affetti da insufficienza pancreatica (1). La prognosi è anche influenzata dal tipo di variante del CFTR, da geni modificatori, dalla microbiologia delle vie aeree, dal sesso, dalla temperatura ambientale, dall'esposizione a inquinanti atmosferici (tra cui il fumo di tabacco), dall'aderenza alle terapie prescritte e dallo stato socio-economico. Il FEV1, corretto per età e sesso, è il miglior indice predittivo della sopravvivenza. Se i risultati della terapia di modulazione del CFTR nella fibrosi cistica sono sostenuti, l'aspettativa di vita può potenzialmente aumentare ulteriormente.
Cure di fine vita
I pazienti e le loro famiglie meritano una discussione delicata alla prognosi e alle preferenze di cura durante tutto il decorso della malattia, specialmente se la funzione polmonare diminuisce progressivamente.
Un segno di rispetto per i pazienti con fibrosi cistica è assicurarsi che vengano fornite loro tutte le informazioni e la possibilità di fare delle scelte, compreso il sostegno nel determinare come e quando accettare l'idea di dover morire.
Quando appropriato, devono essere offerte cure palliative, compresa una sufficiente gestione dei sintomi, per assicurare una pacifica assistenza di fine vita. Una strategia utile da considerare per il paziente è far accettare una prova limitata nel tempo di un trattamento pienamente aggressivo se necessario, concordando prima i parametri che devono indicare quando cessare le cure aggressive (vedi Ordini di non risuscitare e ordini medici portabili).
Riferimenti relativi alla prognosi
1. Cystic Fibrosis Foundation Patient Registry 2021 Annual Data Report Bethesda, Maryland 2022 Cystic Fibrosis Foundation. Consultato il 20/10/2023.
Punti chiave
La fibrosi cistica è causata da 2 varianti del gene per una proteina chiamata cystic fibrosis transmembrane conductance regulator (CFTR), (regolatore della conduttanza transmembrana della fibrosi cistica), che regola il trasporto di cloruro, sodio e bicarbonato attraverso le membrane epiteliali.
Le principali complicanze riguardano i polmoni, con danno a carico delle piccole e grandi vie aeree, infiammazione cronica, e infezioni batteriche croniche e ricorrenti, in particolare da Pseudomonas aeruginosa.
Altre conseguenze importanti comprendono un'insufficienza pancreatica, che porta a malassorbimento di nutrienti e vitamine con conseguente riduzione della crescita e dello sviluppo, e, nei pazienti anziani, al rischio di diabete.
Misure di evacuazione delle vie aeree (p. es., drenaggio posturale, percussione, vibrazioni, tosse assistita), mucolitici e idratatori delle vie aeree sono spesso iniziati nella prima infanzia; è raccomandato un regolare esercizio aerobico.
I farmaci che correggono o potenziano il CFTR (modulatori del CFTR) possono migliorare l'evoluzione dei pazienti che hanno alcune varianti del CFTR.
Gli antibiotici vengono somministrati precocemente in ogni esacerbazione polmonare; la selezione del farmaco può essere basata sulla coltura e sui test di sensibilità agli antibiotici.
La dieta deve essere integrata con enzimi pancreatici, vitamine ad alto dosaggio, e dal 30 al 50% di calorie in più derivanti principalmente dai grassi.
Per ulteriori informazioni
Le seguenti risorse in lingua inglese possono essere utili. Si noti che il Manuale non è responsabile per il contenuto di questa risorsa.
Cystic Fibrosis Foundation: Age-specific care guidelines for cystic fibrosis







