

Dilated cardiomyopathy is myocardial dysfunction causing heart failure in which ventricular dilation and systolic dysfunction predominate. Symptoms include dyspnea, fatigue, and peripheral edema. Diagnosis is clinical and by elevated natriuretic peptides, chest x-ray, echocardiography, and MRI. Treatment is directed at the cause. Standard heart failure treatment measures are used (eg, angiotensin-converting enzyme inhibitors, beta-blockers, aldosterone receptor blockers, angiotensin II receptor blockers, ARNI [angiotensin II receptor blocker and neprilysin inhibitor], sodium glucose cotransporter 2 protein [SGLT2] inhibitors, hydralazine/nitrates, diuretics, digoxin). If heart failure is progressive and severe, cardiac resynchronization therapy, implantable cardioverter-defibrillator, repair of moderate to severe valvular regurgitation, left ventricular assist device, or heart transplantation may be needed.receptor blocker and neprilysin inhibitor], sodium glucose cotransporter 2 protein [SGLT2] inhibitors, hydralazine/nitrates, diuretics, digoxin). If heart failure is progressive and severe, cardiac resynchronization therapy, implantable cardioverter-defibrillator, repair of moderate to severe valvular regurgitation, left ventricular assist device, or heart transplantation may be needed.
A cardiomyopathy is a primary disorder of the heart muscle (see also Overview of Cardiomyopathies). Dilated cardiomyopathy can develop at any age but is more common in adults younger than about 50 years of age. About 10% of people who develop dilated cardiomyopathy are older than 65. In the United States, the disorder occurs in 3 times as many males than in females and in twice as many people of African ancestry compared with White people (1). About 5 to 6 of every 100,000 people develop the disorder each year (1).

General reference
1. Ntusi NAB, Sliwa K. Impact of Racial and Ethnic Disparities on Patients With Dilated Cardiomyopathy: JACC Focus Seminar 7/9. J Am Coll Cardiol 2021;78(25):2580-2588. doi:10.1016/j.jacc.2021.10.021
Pathophysiology of Dilated Cardiomyopathy
As a primary myocardial disorder, the myocardial dysfunction of dilated cardiomyopathy occurs in the absence of other disorders that can cause dilated myocardium, such as severe occlusive coronary artery disease or conditions that involve pressure or volume overload of the ventricle (eg, hypertension, valvular heart disease). In some patients, dilated cardiomyopathy is believed to start with acute myocarditis (probably viral in most cases), followed by a variable latent phase, a phase with diffuse necrosis of myocardial myocytes (due to an autoimmune reaction to virus-altered myocytes), and chronic fibrosis. Regardless of the cause, the myocardium dilates, thins, and hypertrophies in compensation (see figure ), often leading to functional mitral regurgitation and/or tricuspid regurgitation and atrial dilation.
The disorder affects both ventricles in most patients, only the left ventricle (LV) in a few, and only the right ventricle (RV) rarely.
Mural thrombi may form due to stasis of blood once chamber dilation and dysfunction are significant. Cardiac tachyarrhythmias often complicate the acute myocarditis and late chronic dilated phases as may atrioventricular block. Atrial fibrillation commonly occurs as the left atrium dilates.
Etiology of Dilated Cardiomyopathy
Dilated cardiomyopathy has many known and probably many unidentified causes (see table ). More than 20 viruses can cause dilated cardiomyopathy; in temperate zones, coxsackievirus B is most common. In Central and South America, Chagas disease due to Trypanosoma cruzi is the most common infectious cause.
Other causes include prolonged (chronic) tachycardia, HIV infection, toxoplasmosis, thyrotoxicosis, and thiamin deficiency (causing beriberi). Many toxic substances, particularly alcohol, various organic solvents, iron or heavy metal ions, and certain chemotherapeutic agents (eg, doxorubicin, trastuzumab), damage the heart. Frequent ventricular ectopy (> 10,000 ventricular premature beats/day) has been associated with left ventricular systolic dysfunction. , various organic solvents, iron or heavy metal ions, and certain chemotherapeutic agents (eg, doxorubicin, trastuzumab), damage the heart. Frequent ventricular ectopy (> 10,000 ventricular premature beats/day) has been associated with left ventricular systolic dysfunction.
Sudden emotional stress and other hyperadrenergic states can trigger acute dilated cardiomyopathy that is typically reversible (as is that caused by prolonged tachycardia). An example is acute apical ballooning cardiomyopathy (also called takotsubo cardiomyopathy, stress cardiomyopathy, or broken heart syndrome). In this disorder, usually the apex and occasionally other segments of the left ventricle are affected, causing regional wall dysfunction and sometimes focal dilation (ballooning).
Genetic factors play a role in 20 to 35% of cases; > 60 genes and loci have been implicated.
Causes of Dilated Cardiomyopathy
Cause | Examples |
|---|---|
Chronic tachycardia | Frequent ventricular ectopy Uncontrolled atrial fibrillation or other persistent tachyarrhythmias |
Eosinophilic myocarditis | |
Genetic abnormality | Familial disease in 20–30% of patients: autosomal dominant, X-linked, autosomal recessive, or mitochondrial inheritance |
Granulomatous disorders | Granulomatosis with polyangiitis Granulomatous or giant cell myocarditis |
Hereditary neuromuscular and neurologic disorders | |
Infections (acute or chronic) | Bacterial Fungal Helminthic Protozoan Rickettsial Spirochetal Viral (including HIV infection) |
Medications, illicit drugs, and toxins | Anthracyclines Catecholamines Cobalt Cocaine Cyclophosphamide Cyclophosphamide DoxorubicinDoxorubicin Heavy metals Organic solvents Psychotherapeutic medications (tricyclic and quadricyclic antidepressants, phenothiazine) Radiation TrastuzumabTrastuzumab |
Metabolic disorders | Familial storage disorders (eg, Gaucher disease) Nutritional disorders (eg, thiamin deficiency, selenium deficiency, carnitine deficiency, kwashiorkor) Severe obesity Uremia |
Pregnancy (peripartum period) | — |
Systemic rheumatic diseases | |
Tumors | Certain endocrinologically active tumors (eg, pheochromocytoma, adrenal tumors, thyroid tumors) |
Symptoms and Signs of Dilated Cardiomyopathy
Onset of dilated cardiomyopathy is usually gradual except in acute myocarditis, acute apical ballooning cardiomyopathy, and tachyarrhythmia-induced cardiomyopathy. About 25% of all patients with dilated cardiomyopathy have atypical chest pain. Other symptoms depend on which ventricle is affected.
Left ventricular dysfunction causes exertional dyspnea and fatigue due to elevated left ventricular diastolic pressure and low cardiac output.
Right ventricular failure causes peripheral edema and neck vein distention. Infrequently the right ventricle is predominantly affected in younger patients, and atrial arrhythmias and sudden death due to malignant ventricular tachyarrhythmias are typical.
Diagnosis of Dilated Cardiomyopathy
Chest x-ray
ECG
Echocardiography
Cardiac MRI
Endomyocardial biopsy (selected cases)
Testing for cause as indicated
Diagnosis of dilated cardiomyopathy is by history, physical examination, and exclusion of other common causes of ventricular failure (eg, systemic hypertension, primary valvular disorders, myocardial infarction—see table ). Particularly in cases of dilated cardiomyopathy without a clear cause, a careful family history should be taken to identify family members with possible early-onset heart disease, heart failure, or sudden death. In many centers, first-degree family members are screened for cardiac dysfunction (such as with echocardiography). Because other common causes of ventricular failure must be excluded, chest x-ray, ECG, echocardiography, and cardiac MRI are required. Endomyocardial biopsy is done in selected cases.
Serum cardiac biomarkers are measured if acute symptoms or chest pain is present. Although typically indicative of coronary ischemia, troponin elevation often occurs in heart failure, especially if renal function is decreased. Serum natriuretic peptide levels are typically elevated when heart failure is present.
Specific causes suspected clinically are diagnosed (see elsewhere in THE MANUAL). If no specific cause is clinically apparent, serum ferritin and iron-binding capacity and thyroid-stimulating hormone levels are measured.
Serologic tests for Toxoplasma, T. cruzi, coxsackievirus, HIV, and echovirus may be done in appropriate cases.
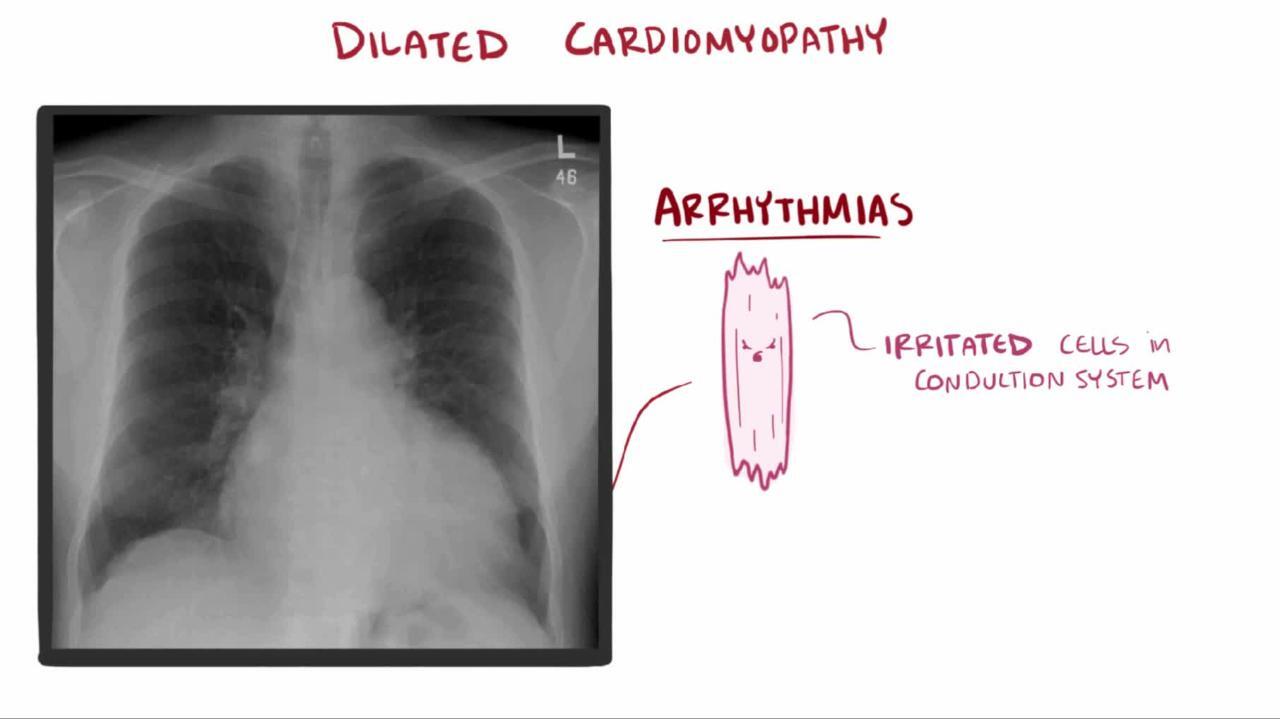
Chest x-ray shows cardiomegaly, usually of all chambers. Pleural effusion, particularly on the right, often accompanies increased pulmonary venous pressure and interstitial edema.

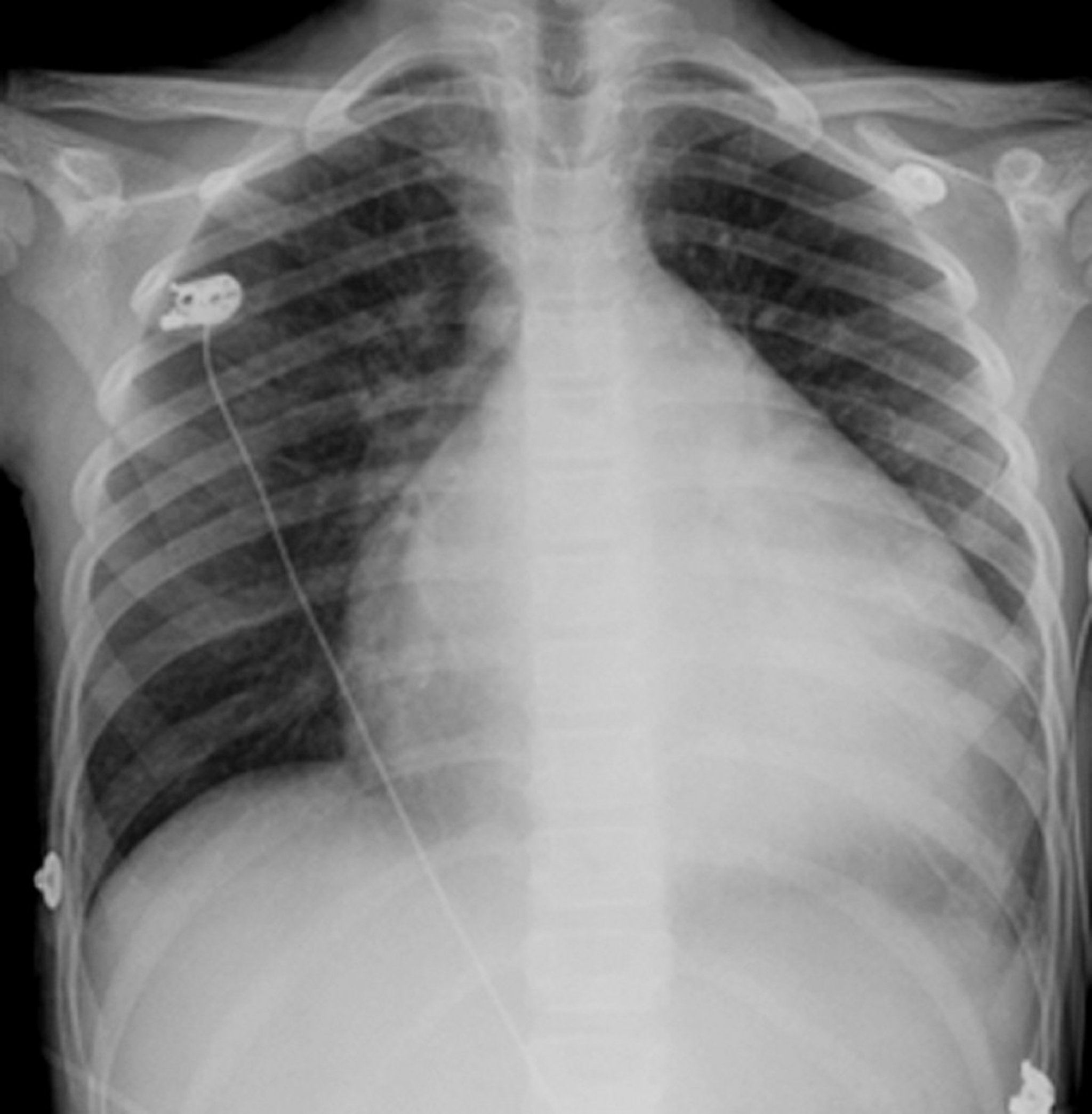
© Springer Science+Business Media
The ECG may show sinus tachycardia and nonspecific ST-segment depression with low voltage or inverted T waves. Sometimes pathologic Q waves are present in the precordial leads, simulating previous myocardial infarction. Left bundle branch block and atrial fibrillation are common.
Echocardiography shows dilated, hypokinetic cardiac chambers and rules out primary valvular disorders. Segmental wall motion abnormalities can also occur in dilated cardiomyopathy because the process may be patchy. Echocardiography may also show a mural thrombus.
Cardiac MRI is useful in providing detailed imaging of myocardial structure and function. MRI with gadolinium contrast may show abnormal myocardial tissue texture or scarring pattern (ie, late gadolinium enhancement, or LGE). The pattern of LGE can be diagnostic in active myocarditis, sarcoidosis, muscular dystrophy, or Chagas disease).

© 2017 Elliot K. Fishman, MD.
Positron-emission tomography (PET) has been shown to be sensitive for diagnosis of cardiac sarcoidosis.
Coronary angiography may be required to exclude coronary artery disease as the cause of LV dysfunction when the diagnosis is in doubt after noninvasive tests. Patients with chest pain or several cardiovascular risk factors and older patients are more likely to have coronary artery disease. Biopsy of either ventricle can be done during catheterization in select cases where the results will change management.
Endomyocardial biopsy is indicated if giant cell myocarditis, eosinophilic myocarditis, or sarcoidosis is suspected, as the results will affect management.
Treatment of Dilated Cardiomyopathy
Cause (if any) treated
Standard therapy for heart failure with reduced ejection fraction
Anticoagulants when atrial fibrillation or other indication is present
Sometimes implantable cardioverter-defibrillator, cardiac resynchronization therapy (CRT), left ventricular assist device, or transplantation
Immunosupression in patients with giant cell myocarditis, eosinophilic myocarditis, sarcoidosis, or other autoimmune diseases (eg, systemic lupus erythematosus)
Treatable causes (eg, toxoplasmosis, acute Chagas disease, hemochromatosis, thyrotoxicosis, thiamin deficiency) are corrected. Patients with HIV infection should have antiretroviral therapy (ART) optimized. Treatment with immunosuppression should be limited to patients with biopsy-proven giant cell myocarditis, eosinophilic myocarditis, sarcoidosis, or other autoimmune causes (eg, systemic lupus erythematosus).
Otherwise, treatment is the same as for heart failure with reduced ejection fraction: angiotensin-converting enzyme (ACE) inhibitors, beta-blockers, aldosterone receptor blockers, angiotensin II receptor blockers, ARNI (angiotensin II receptor blocker and neprilysin inhibitor), sodium glucose cotransporter 2 protein (SGLT2) inhibitors, hydralazine/nitrates, diuretics, and digoxin. Older studies have suggested that patients with idiopathic dilated cardiomyopathy respond particularly well to standard heart failure treatments and generally do better than patients with ischemic heart disease, but more recent data do not support this difference, especially in view of changes in drug regiments over the years (receptor blocker and neprilysin inhibitor), sodium glucose cotransporter 2 protein (SGLT2) inhibitors, hydralazine/nitrates, diuretics, and digoxin. Older studies have suggested that patients with idiopathic dilated cardiomyopathy respond particularly well to standard heart failure treatments and generally do better than patients with ischemic heart disease, but more recent data do not support this difference, especially in view of changes in drug regiments over the years (1).
Special precautions are needed in the treatment of peripartum cardiomyopathy. Many medications (eg, ACE inhibitors and angiotensin II receptor blockers) should be avoided during pregnancy due to the risk of fetal harm. In addition, these medications are not recommended for women who are breastfeeding.
Prophylactic oral anticoagulation has been used in the past to prevent mural thrombus in other forms of cardiomyopathy. The use of anticoagulants for patients with reduced left ventricular function and in sinus rhythm remains controversial, and anticoagulant use in this situation is not routine. Warfarin or a direct oral anticoagulant (DOAC) is recommended when a specific indication is present (eg, previous cerebrovascular embolism, identified cardiac thrombus, atrial fibrillation, atrial flutter). has been used in the past to prevent mural thrombus in other forms of cardiomyopathy. The use of anticoagulants for patients with reduced left ventricular function and in sinus rhythm remains controversial, and anticoagulant use in this situation is not routine. Warfarin or a direct oral anticoagulant (DOAC) is recommended when a specific indication is present (eg, previous cerebrovascular embolism, identified cardiac thrombus, atrial fibrillation, atrial flutter).
Both the American Heart Association and European Society of Cardiology Guidelines recommend considering anticoagulation therapy in patients with peripartum cardiomyopathy who have very low ejection fractions given the risk of a hypercoagulable state during pregnancy (2, 3). Low molecular weight heparin has been used. Warfarin and direct-acting oral anticoagulants, however, should not be used during certain stages of pregnancy due to fetal risk.). Low molecular weight heparin has been used. Warfarin and direct-acting oral anticoagulants, however, should not be used during certain stages of pregnancy due to fetal risk.
Medical treatment of heart failure reduces risk of arrhythmia, but an implantable cardioverter-defibrillator may be used to prevent death due to sudden arrhythmia in patients who continue to have a reduced ejection fraction despite optimal medical therapy. Because atrioventricular (AV) block during acute myocarditis often resolves, a permanent pacemaker is usually not needed acutely. However, a permanent pacemaker may be required if AV block persists or develops during the chronic dilated phase. If patients have a widened QRS interval with a low left ventricular ejection fraction and severe symptoms despite optimal medical treatment, cardiac resynchronization therapy should be considered.
Patients with refractory heart failure despite treatment may become candidates for heart transplantation. Selection criteria include absence of associated systemic disorders and psychologic disorders and high, irreversible pulmonary vascular resistance; because donor hearts are scarce, younger patients (usually < 70 years) are given higher priority. Left ventricular assist devices (LVAD) may also be considered as a bridge to heart transplantation or as destination therapy in some patients (eg, patients who are not eligible for heart transplantation).
Treatment references
1. Bozkurt B, Colvin M, Cook J, et al: Current Diagnostic and Treatment Strategies for Specific Dilated Cardiomyopathies: A Scientific Statement From the American Heart Association [published correction appears in Circulation 134(23):e652, 2016]. Circulation 134(23):e579–e646, 2016. doi:10.1161/CIR.0000000000000455
2. Bozkurt B, Colvin M, Cook J, et al: Current diagnostic and treatment strategies for specific dilated cardiomyopathies: A Scientific Statement From the American Heart Association. Circulation 134(23):e579–e646.2, 2016. doi:10.1161/CIR.0000000000000455
3. Regitz-Zagrosek V, Roos-Hesselink JW, Bauersachs J, et al: 2018 ESC Guidelines for the management of cardiovascular diseases during pregnancy: The Task Force for the Management of Cardiovascular Diseases during Pregnancy of the European Society of Cardiology (ESC). Eur Heart J 39: 3165–3241, 2018. doi: 10.1093/eurheartj/ehy340
Prognosis for Dilated Cardiomyopathy
Prognosis for dilated cardiomyopathy generally has been poor, although prognosis has improved with current management regimens (eg, use of beta-blockers, angiotensin-converting enzyme [ACE] inhibitors, angiotensin II receptor blockers [ARB], angiotensin II receptor blocker and neprilysin inhibitor [ARNI], mineralocorticoid receptor antagonists, sodium glucose cotransporter 2 protein (SGLT2) inhibitors, implantable cardioverter-defibrillators, or cardiac resynchronization therapy). About 20% of patients die in the first year and then about 10%/year thereafter; about 40 to 50% of deaths are sudden, due to a malignant arrhythmia (1). Prognosis is better for females than for males, and people of African ancestry survive about half as long as White people (1).
Prognosis is better if compensatory hypertrophy preserves ventricular wall thickness and is worse if ventricular walls thin markedly and the ventricle dilates. Patients whose dilated cardiomyopathy is well-compensated with treatment may be stable for many years.
Prognosis reference
1. Schultheiss HP, Fairweather D, Caforio ALP, et al. Dilated cardiomyopathy. Nat Rev Dis Primers 2019;5(1):32. doi:10.1038/s41572-019-0084-1
2.
Key Points
In dilated cardiomyopathy, the myocardium dilates, thins, and hypertrophies.
Causes include infection (commonly viral), toxins, and metabolic, genetic, or systemic rheumatic disorders.
Do chest x-ray, ECG, echocardiography, and cardiac MRI to evaluate extent of disease and endomyocardial biopsy in selected patients.
Look for other causes of heart failure.
Treat primary cause if possible and use standard heart failure treatment measures (eg, angiotensin-converting enzyme [ACE] inhibitors, beta-blockers, aldosterone receptor blockers, angiotensin II receptor blockers, ARNI [angiotensin II receptor blocker and neprilysin inhibitor], sodium glucose cotransporter 2 protein (SGLT2) inhibitors, hydralazine/nitrates, diuretics, digoxin, implantable cardioverter-defibrillator, and/or cardiac resynchronization therapy).receptor blocker and neprilysin inhibitor], sodium glucose cotransporter 2 protein (SGLT2) inhibitors, hydralazine/nitrates, diuretics, digoxin, implantable cardioverter-defibrillator, and/or cardiac resynchronization therapy).
Use oral anticoagulants and immunosuppressants in selected patients.
More Information
The following English-language resources may be useful. Please note that THE MANUAL is not responsible for the content of these resources.
American Heart Association guidelines on dilated cardiomyopathies: Bozkurt B, Colvin M, Cook J, et al: Current diagnostic and treatment strategies for specific dilated cardiomyopathies: A Scientific Statement From the American Heart Association. Circulation 134(23):e579–e646.2, 2016. doi:10.1161/CIR.0000000000000455
European Society of Cardiology guidelines on cardiomyopathy in pregnant patients: Regitz-Zagrosek V, Roos-Hesselink JW, Bauersachs J, et al: 2018 ESC Guidelines for the management of cardiovascular diseases during pregnancy: The Task Force for the Management of Cardiovascular Diseases during Pregnancy of the European Society of Cardiology (ESC). European Heart Journal 39: 3165–3241, 2018. doi: 10.1093/eurheartj/ehy340





