

Sickle cell disease (a hemoglobinopathy) causes a chronic hemolytic anemia occurring commonly in people with African ancestry. Sickle cell anemia is caused by homozygous inheritance of genes for hemoglobin (Hb) S or Hb S beta 0 thalassemia. Sickle cell disease includes individuals with sickle cell anemia as well as those with HbSC disease, HbS beta + thalassemia, and other rare sickle cell disease causing variants. Sickle-shaped red blood cells (RBCs) cause vaso-occlusion and are prone to hemolysis, leading to severe pain crises, organ ischemia, and other systemic complications. Acute exacerbations (crises) may develop frequently. Infection, bone marrow aplasia, or lung involvement (acute chest syndrome) can develop acutely and be fatal. Anemia is present, and sickle cells are usually evident on the peripheral smear. Diagnosis requires hemoglobin electrophoresis. Painful crises are treated with analgesics and other supportive measures. Transfusions are often given acutely when neurovascular complications, acute chest syndrome, or other multiorgan failure occurs. Long-term prophylactic transfusion therapy may be indicated but may be limited depending on local blood supply availability and history of alloimmunization or hemolytic transfusion reactions. Vaccines against bacterial infections, prophylactic antibiotics, and aggressive treatment of infections prolong survival. Hydroxyurea can decrease the frequency of crises and the acute chest syndrome. Use of curative therapies including bone marrow transplant and gene therapy is expanding.
Hemoglobin S was the first abnormal hemoglobin to be identified.
Patients who are homozygous for HbS (approximately 0.3% of people with African ancestry in the United States) have sickle cell anemia; patients who are heterozygous, called sickle cell trait (about 8% of people with African ancestry in the United States), are not anemic but have a risk of other complications (1). In 2021, an estimated 7.74 million people were living with sickle-cell disease globally (2).
General references
1. Centers for Disease Control and Prevention: Sickle cell disease (SCD): Data and statistics on sickle cell disease. May 15, 2024. Accessed February 6, 2026.
2. World Health Organization. Sickle-cell disease: Key Facts. August 6, 2025. Accessed February 13, 2026.
Pathophysiology of Sickle Cell Disease
In hemoglobin S, valine is substituted for glutamic acid in the sixth amino acid of the beta chain. Deoxygenated Hb S is much less soluble than oxygenated Hb A; it polymerizes and forms a semisolid gel that causes RBCs to deform into a sickle shape at sites of low partial pressure of oxygen (PO2). Distorted, inflexible RBCs adhere to vascular endothelium, cause inflammation and vasoconstriction, and plug small arterioles and capillaries, which leads to infarction of both the microvasculature and large vessels (eg, causing stroke). Vaso-occlusion also causes endothelial injury, which results in inflammation and can lead to thromboses. Because sickled RBCs are fragile, the mechanical trauma of circulation causes hemolysis (see Overview of Hemolytic Anemia). Chronic compensatory bone marrow hyperactivity and ischemic damage deform the bones.
Acute exacerbations
Acute exacerbations (crises) occur intermittently, often for no known reason. In some cases, crisis appears to be precipitated by:
Fever
Viral infection
Local trauma
Vaso-occlusive crisis (pain crisis) is the most common type; it is caused by tissue hypoxia and leads to ischemia and infarction, typically in the bones, but also in the spleen, lungs, or kidneys.
Aplastic crisis occurs when bone marrow erythropoiesis slows during acute infection due to human parvovirus, during which an acute erythroblastopenia may occur.
Acute chest syndrome results from pulmonary microvascular occlusion and is a common cause of death. It occurs in all age groups but is most common in childhood. Repeated episodes as well as other factors predispose to chronic pulmonary hypertension.
Acute multisystem organ failure is a life-threatening complication that rapidly affects multiple organ systems. It often first presents as severe acute vaso-occlusive crisis or acute chest syndrome and often thrombocytopenia and anemia with reticulocytopenia. It is thought to be driven by fat embolization.
Sequestration crisis typically occurs in children whose spleen has not yet become fibrotic due to repeated splenic infarction. Acute sequestration of sickled cells in the spleen exacerbates anemia. This is accompanied by at least a 2 g/dL (20 g/L) fall in hemoglobin and may lead to hemodynamic instability.
Hepatic sequestration may occur in children or adults, causing right upper quadrant pain, rapid enlargement of the liver, and at least a 2 g/dL (20 g/L) decrease in hemoglobin.
Intrahepatic cholestasis causes right upper quadrant tenderness, hepatomegaly, marked hyperbilirubinemia, coagulopathy, and often renal failure. It is a result of disseminated vaso-occlusion in the hepatic sinusoids leading to liver ischemia.
Priapism, a serious complication that can cause erectile dysfunction, is most common in young males.
Complications
Chronic spleen damage can lead to autoinfarction and increases susceptibility to infection, particularly pneumococcal and Salmonella infections (including Salmonella osteomyelitis). These infections are especially common in early childhood and can be fatal.
Recurrent ischemia and infarction can cause chronic dysfunction of multiple different organ systems. Complications include ischemic stroke, moyamoya, seizures, avascular necrosis, renal concentrating defects, renal papillary necrosis, chronic kidney disease, heart failure, pulmonary hypertension, pulmonary fibrosis, and retinopathy.
Heterozygotes
Patients who are heterozygous (Hb AS) do not experience hemolysis or painful crises. However, they do have an increased risk of chronic kidney disease and pulmonary embolism (1). In addition, rhabdomyolysis may occur rarely during sustained, exhausting exercise. Impaired ability to concentrate urine (hyposthenuria) is common. Unilateral hematuria (by unknown mechanisms and usually from the left kidney) can occur but is self-limited. Typical renal papillary necrosis can occur but is less common than among patients who are homozygous, and there is an association with the extremely rare medullary carcinoma of the kidney.
Pathophysiology reference
1. Naik RP, Smith-Whitley K, Hassell KL, et al. Clinical Outcomes Associated With Sickle Cell Trait: A Systematic Review. Ann Intern Med. 2018;169(9):619-627. doi:10.7326/M18-1161
Symptoms and Signs of Sickle Cell Disease
Most symptoms result from:
Anemia
Vaso-occlusive events resulting in tissue ischemia, infarction, and inflammation
Anemia is can be severe but varies among patients and genotypes and is usually compensated; mild jaundice and pallor are common.
Hepatosplenomegaly is common in children, but because of repeated infarctions and subsequent fibrosis (autosplenectomy), the spleen in adults is commonly atrophied. Cardiomegaly and systolic ejection (flow) murmurs are common. Cholelithiasis and chronic punched-out skin ulcers around the ankles occur.
Painful vaso-occlusive crisis causes severe pain in long bones, the hands and feet, back, and joints. Chronically, vaso-occlusion can lead to avascular necrosis of bone.
Acute chest syndrome is characterized by sudden onset of fever, chest pain, and pulmonary infiltrates. It may follow bacterial pneumonia. Hypoxemia may develop rapidly, causing dyspnea.
Diagnosis of Sickle Cell Disease
DNA testing (prenatal diagnosis or sometimes after birth if diagnosis is unclear)
Peripheral blood smear
Solubility testing
Hemoglobin electrophoresis,thin-layer isoelectric focusing, or high performance liquid chromatography
Patients with symptoms or signs suggesting the disorder or its complications (eg, poor growth, acute and unexplained bone pain, particularly in the fingers, aseptic necrosis of the femoral head, unexplained hematuria), and patients with African ancestry and normocytic anemia (particularly if hemolysis is present) require laboratory tests for hemolytic anemia, hemoglobin electrophoresis, and examination of RBCs for sickling. Cells are normocytic (microcytosis suggests a concomitant alpha or beta thalassemia). Nucleated RBCs frequently appear in the peripheral blood, and reticulocytosis ≥ 10% is common. Dry-stained smears may show sickled RBCs (crescent-shaped, often with elongated or pointed ends).

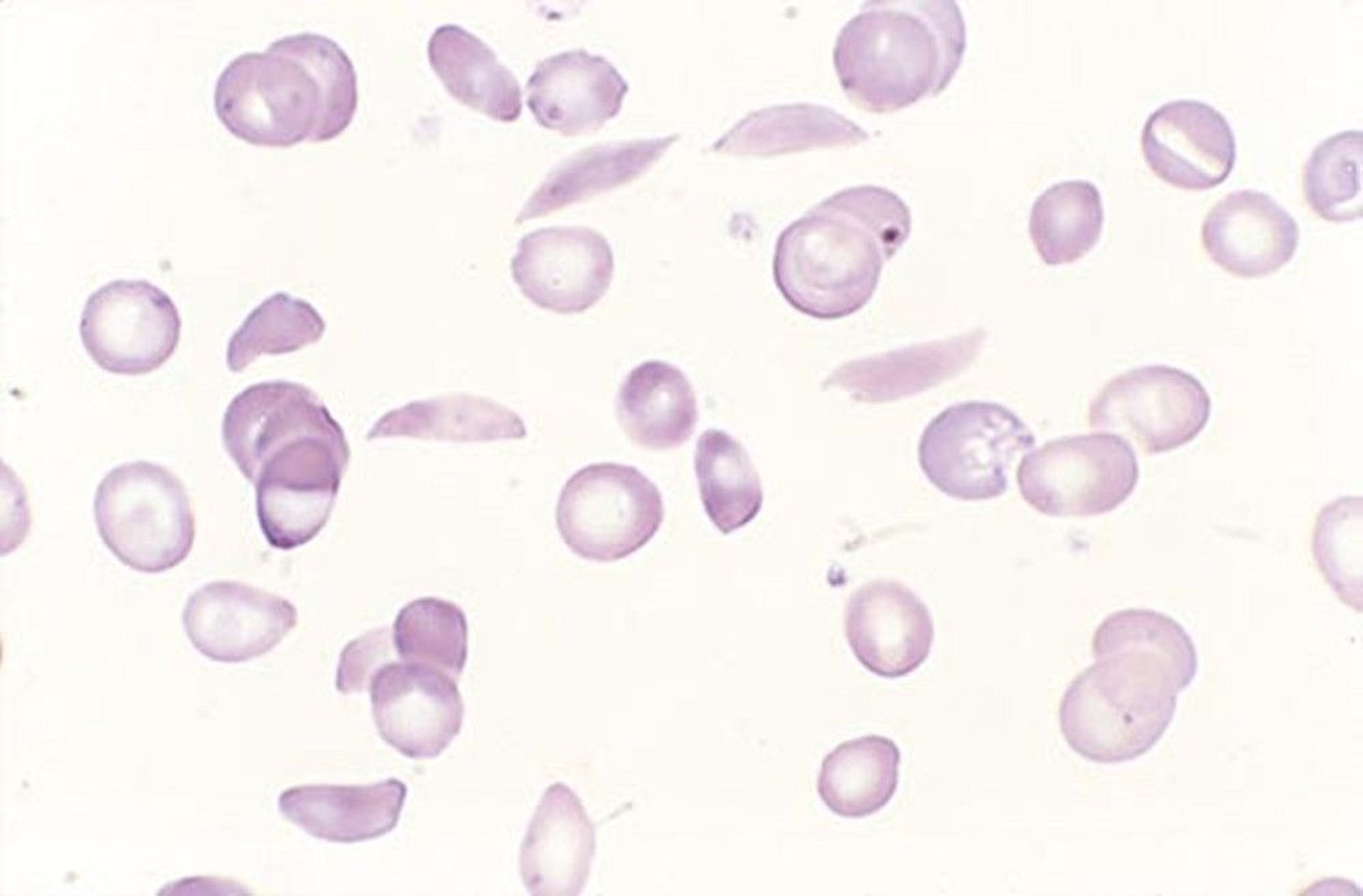
By permission of the publisher. From Tefferi A, Li C. In Atlas of Clinical Hematology. Edited by JO Armitage. Philadelphia, Current Medicine, 2004.
The homozygous state is differentiated from other hemoglobinopathies by electrophoresis showing only Hb S with a variable amount of Hb F. The heterozygous state is differentiated by the presence of more Hb A than Hb S on electrophoresis.
Bone marrow examination is not used for diagnosis. If it is done to differentiate other anemias, it shows hyperplasia, with normoblasts predominating; bone marrow may become aplastic during severe infections or crisis. The erythrocyte sedimentation rate, if done to exclude other disorders (eg, juvenile rheumatoid arthritis causing hand and foot pain), is low.
Incidental findings on skeletal radiographs may include widening of the diploic spaces of the skull and a sun-ray appearance of the diploic trabeculations. The long bones often show cortical thinning, irregular densities, and new bone formation within the medullary canal.
Unexplained hematuria, even among patients not suspected of having sickle cell disease, should prompt consideration of sickle cell trait.
Evaluation of exacerbations
If patients with known sickle cell disease have acute exacerbations, including pain, fever, or other symptoms of infection, a complete blood count, reticulocyte count, and comprehensive metabolic panel are done. A reticulocyte count < 1% suggests aplastic crisis, particularly when hemoglobin decreases below the patient’s usual level. In a painful crisis without aplasia, the white blood cell count rises, often with a shift to the left, particularly during bacterial infection. The platelet count is usually increased but can fall with the acute chest syndrome. If measured, serum bilirubin is usually elevated (eg, 2 to 4 mg/dL [34 to 68 micromol/L]), and urine may contain urobilinogen. Vaso-occlusive crisis is a clinical diagnosis and cannot be confirmed by specific laboratory findings, though certain lab findings may suggest the severity of the presentation and risk for decompensation (eg, thrombocytopenia, reticulocytopenia, elevation in liver function tests or creatinine, and troponin elevation).
In patients with chest pain or difficulty breathing, acute chest syndrome and pulmonary embolism are considered; chest radiographs and pulse oximetry are necessary. Because acute chest syndrome is the leading cause of death in sickle cell disease, early recognition and treatment usually with blood transfusions are critical. Hypoxemia or pulmonary parenchymal infiltrates on chest radiographs suggest acute chest syndrome or pneumonia. New hypoxemia without pulmonary infiltrates may suggest pulmonary embolism.
In patients with fever, infection and acute chest syndrome are considered; cultures, chest radiograph, and other diagnostic tests based on the patient's symptoms are done.
Screening for Sickle Cell Disease
The type of testing done depends on the age of the patient. DNA testing can be used for prenatal diagnosis or to confirm a diagnosis of the sickle cell genotype. Screening of neonates involves hemoglobin electrophoresis. Screening in children and adults involves examination of the peripheral smear, hemoglobin solubility testing, and hemoglobin electrophoresis or high performance liquid chromatography (HPLC).
Prenatal screening
PCR (polymerase chain reaction) is sensitive when used for prenatal diagnosis. It is recommended for families at risk for sickle cell (eg, couples with medical or family histories of anemia or of suggestive ethnic background). DNA samples can be obtained by chorionic villus sampling at 10 to 12 weeks gestation. Amniotic fluid can also be tested at 14 to 16 weeks. Diagnosis is important for genetic counseling.
Newborn screening
Universal testing is recommended. Testing for sickle cell disease is frequently one of a battery of newborn screening tests. To distinguish between hemoglobin (Hb) F, Hb S, Hb A, and Hb C, the recommended tests are hemoglobin electrophoresis using cellulose acetate or acid citrate agar, thin-layer isoelectric focusing, or hemoglobin fractionation by HPLC. Repeat testing at age 3 to 6 months may be necessary for confirmation. Solubility testing for Hb S is unreliable during the first few months of life.
Screening of children and adults
Patients with a family history of sickle cell disease or trait should be screened with peripheral smear, hemoglobin (Hb) solubility testing, and hemoglobin electrophoresis or HPLC.
Treatment of Sickle Cell Disease
Broad-spectrum antibiotics (for infection)
Analgesics and IV hydration (for vaso-occlusive pain crisis)
Oxygen (for hypoxia)
Sometimes transfusions
Immunizations, folic acid supplementation, and hydroxyurea (for prevention of disease complications and exacerbations)Immunizations, folic acid supplementation, and hydroxyurea (for prevention of disease complications and exacerbations)
Stem cell transplantation or gene therapy (for advanced complications)
Treatment includes regular health maintenance measures as well as specific treatment of the complications as they arise. Complications are treated supportively.
Indications for hospitalization include suspected serious (including systemic) infection, aplastic crisis, acute chest syndrome, and, often, intractable pain. Fever alone may not be a reason to hospitalize. However, patients who appear acutely ill and have a temperature > 38° C should be admitted so that cultures can be obtained and IV antibiotics can be given.
Antibiotics
Patients with suspected serious bacterial infections or acute chest syndrome require broad-spectrum antibiotics immediately.
Analgesics
Painful crises are managed with liberal administration of analgesics, usually opioids. Meperidine should be used with caution. Nonsteroidal anti-inflammatory drugs (NSAIDs) are often useful in reducing opioid requirements; however, they must be used cautiously in patients with renal disease. Painful crises are managed with liberal administration of analgesics, usually opioids. Meperidine should be used with caution. Nonsteroidal anti-inflammatory drugs (NSAIDs) are often useful in reducing opioid requirements; however, they must be used cautiously in patients with renal disease.
Intravenous hydration
Although dehydration contributes to sickling and may precipitate crises, it is unclear whether vigorous hydration is helpful during crises. Nevertheless, maintaining normal intravascular volume has been a mainstay of therapy.
Oxygen
Oxygen is given if needed to treat hypoxia.
Transfusion
Transfusion is given in many situations in which its efficacy has not been systematically studied. However, chronic transfusion therapy is indicated for prevention of recurrent cerebral thrombosis, especially in children, in an effort to maintain the Hb S percentage less than 30% (1).
In the acute setting, specific indications for transfusion include:
Acute splenic sequestration with symptomatic anemia
Aplastic crises
Cardiopulmonary symptoms or signs (eg, high-output heart failure, hypoxemia with PO2 < 65 mm Hg)
Preoperative use
Priapism
Life-threatening events that would benefit from improved oxygen delivery, including sepsis, severe infection, acute chest syndrome, acute stroke, and acute organ ischemia (eg, multiorgan system failure, intrahepatic cholestasis)
Sometimes pregnancy
Transfusion is not helpful during an uncomplicated painful crisis.
Simple transfusion can be done when the goal is to correct anemia, such as during aplastic crisis. Transfusion in patients with acute splenic or liver sequestration and symptomatic anemia should be performed with caution because blood sequestered in these organs can reenter the circulation and cause hyperviscosity, which can present with neurologic complications.
Exchange transfusion is performed during severe acute events such as the acute chest syndrome or stroke in order to decrease the Hb S percentage and prevent ischemia. If the initial hemoglobin is low, an exchange transfusion cannot be initiated before first transfusing red cells. Partial exchange transfusion minimizes iron accumulation and hyperviscosity.
Curative treatments
Hematopoietic stem cell transplantation, traditionally with a matched sibling donor and increasingly using alternative donors, remains the only curative treatment for sickle cell disease (2). It is generally restricted to patients with complications (eg, frequent or increasing vaso-occlusive episodes, history of severe or recurrent acute chest syndrome, progressive organ involvement, strokes) (3).
Gene therapy or gene editing techniques that increase the amount of Hb F are available. This field is rapidly evolving, and use of stem cell therapy to treat sickle cell disease will likely continue to expand (4, 5). There are currently 2 available gene therapies for patients 12 years and older utilizing autologous transplantation of hematopoietic stem and progenitor cells. Indications include history of severe vaso-occlusive complications; however, patients with a history of stroke or significant organ damage were excluded from trials. Lovotibeglogene autotemcel is a lentiviral gene therapy that leads to the production of a beta globin variant (HbA T87Q) that prevents sickling (). There are currently 2 available gene therapies for patients 12 years and older utilizing autologous transplantation of hematopoietic stem and progenitor cells. Indications include history of severe vaso-occlusive complications; however, patients with a history of stroke or significant organ damage were excluded from trials. Lovotibeglogene autotemcel is a lentiviral gene therapy that leads to the production of a beta globin variant (HbA T87Q) that prevents sickling (4). Exagamglogene autotemcel uses CRISPR-Cas9 gene editing to reactivate fetal hemoglobin production (). Exagamglogene autotemcel uses CRISPR-Cas9 gene editing to reactivate fetal hemoglobin production (5).
Pharmacologic treatment
Supplemental folic acid, 1 mg orally once a day, is usually prescribed.Supplemental folic acid, 1 mg orally once a day, is usually prescribed.
Hydroxyurea, by increasing Hb F and thereby reducing sickling, decreases painful crises (eg, by approximately 45% [Hydroxyurea, by increasing Hb F and thereby reducing sickling, decreases painful crises (eg, by approximately 45% [6, 7]); it decreases acute chest syndrome and transfusion requirements (7). It is recommended in all children and generally continued through adulthood. In adults not already taking hydroxyurea, it is indicated in patients with recurrent pain crises or other complications. The dose of hydroxyurea is variable and is adjusted based on blood counts and adverse effects. Hydroxyurea causes neutropenia and thrombocytopenia. Its teratogenic effect is debated, and current recommendations advise it should not be given to females during pregnancy or pregnancy planning except in select high-risk instances (8, 9).
Two additional medications are available to treat sickle cell disease. Both L-glutamine and crizanlizumab target vaso-occlusion and were found to decrease pain crises in controlled, randomized studies (Two additional medications are available to treat sickle cell disease. Both L-glutamine and crizanlizumab target vaso-occlusion and were found to decrease pain crises in controlled, randomized studies (10, 11). L-Glutamine is thought to reduce oxidative stress in sickle cells, while crizanlizumab inhibits P-selectin, which contributes to adhesion of sickle cells to the vascular endothelium. While these medications are incorporated into treatment regimens for patients with sickle cell disease, data on their efficacy are limited and conflicting (12).
Ongoing management
Infections due to functional asplenia lead to significantly increased morbidity and mortality, and appropriate immunization, monitoring, and antibiotic prophylaxis are recommended. For long-term management, the following interventions have reduced mortality, particularly during childhood (13):
Pneumococcal, Haemophilus influenzae (inactivated, not live), and meningococcal vaccines. COVID-19 and influenza vaccinations are also recommended. RSV vaccination may be beneficial in adults over 60 years.
Early identification and treatment of serious bacterial infections
Prophylactic antibiotics, including continuous prophylaxis with oral penicillin from birth to at least 5 years
Continuation of hydroxyurea (Continuation of hydroxyurea (14, 15)
Transcranial Doppler flow studies in children can help predict risk of stroke, and many experts recommend annual screening for children from age 2 to 16 years. Children at high risk appear to benefit from prophylactic, chronic exchange transfusions to keep Hb S at < 30% of total hemoglobin (16, 17); iron overload is common and must be screened for and treated.
For patients receiving frequent red cell transfusions, chelation therapy to prevent or delay complications due to iron overload should be considered.
Treatment references
1. DeBaun MR, Jordan LC, King AA, et al. American Society of Hematology 2020 guidelines for sickle cell disease: prevention, diagnosis, and treatment of cerebrovascular disease in children and adults. Blood Adv. 2020;4(8):1554-1588. doi:10.1182/bloodadvances.2019001142
2. Kassim AA, Walters MC, Eapen M, et al. Haploidentical Bone Marrow Transplantation for Sickle Cell Disease. NEJM Evid. 2025;4(3):EVIDoa2400192. doi:10.1056/EVIDoa2400192
3. Kanter J, Liem RI, Bernaudin F, et al. American Society of Hematology 2021 guidelines for sickle cell disease: stem cell transplantation. Blood Adv. 2021;5(18):3668-3689. doi:10.1182/bloodadvances.2021004394C
4. Kanter J, Walters MC, Krishnamurti L, et al. Biologic and Clinical Efficacy of LentiGlobin for Sickle Cell Disease. N Engl J Med. 2022;386(7):617-628. doi:10.1056/NEJMoa2117175
5. Frangoul H, Locatelli F, Sharma A, et al. Exagamglogene Autotemcel for Severe Sickle Cell Disease. N Engl J Med. 2024;390(18):1649-1662. doi:10.1056/NEJMoa2309676
6. Charache S, Terrin ML, Moore RD, et al: Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N Engl J Med. 1995;332(20):1317-1322. doi:10.1056/NEJM199505183322001
7. Rankine-Mullings AE, Nevitt SJ: Hydroxyurea (hydroxycarbamide) for sickle cell disease. Cochrane Database Syst Rev. 2022;9(9):CD002202. doi:10.1002/14651858.CD002202.pub3
8. Habibi A, Etienne-Julan M, Dimopoulou M, et al. Outcomes of pregnancies in sickle cell patients treated with hydroxyurea : Findings from the escort-HU cohort studies. . Outcomes of pregnancies in sickle cell patients treated with hydroxyurea : Findings from the escort-HU cohort studies.Blood. 2025;146 (Supplement 1):2. doi: 10.1182/blood-2025-2
9. Kroner BL, Hankins JS, Pugh N, et al. Pregnancy outcomes with hydroxyurea use in women with sickle cell disease. Am J Hematol. 2022;97(5):603-612. doi:10.1002/ajh.26495
10. Ataga KI, Kutlar A, Kanter J, et al: Crizanlizumab for the prevention of pain crises in sickle cell disease. N Engl J Med. 2017;376(5):429-439. doi: 10.1056/NEJMoa1611770
11. Niihara Y, Miller ST, Kanter J, et al: A phase 3 trial of l-glutamine in sickle cell disease. N Engl J Med 379(3):226–235, 2018. doi: 10.1056/NEJMoa1715971
12. Abboud MR, Cançado RD, De Montalembert M, et al. Crizanlizumab with or without hydroxyurea in patients with sickle cell disease (STAND): primary analyses from a placebo-controlled, randomised, double-blind, phase 3 trial. Lancet Haematol. 2025;12(4):e248-e257. doi:10.1016/S2352-3026(24)00384-3
13. Riddington C, Owusu-Ofori S. Prophylactic antibiotics for preventing pneumococcal infection in children with sickle cell disease. Cochrane Database Syst Rev. 2002;(3):CD003427. doi:10.1002/14651858.CD003427
14. Steinberg MH, Barton F, Castro O, et al. Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: risks and benefits up to 9 years of treatment. JAMA. 2003;289(13):1645-1651. doi:10.1001/jama.289.13.1645
15. Steinberg MH, McCarthy WF, Castro O, et al. The risks and benefits of long-term use of hydroxyurea in sickle cell anemia: A 17.5 year follow-up. Am J Hematol. 2010;85(6):403-408. doi:10.1002/ajh.21699
16. Biller E, Zhao Y, Berg M, et al. Red blood cell exchange in patients with sickle cell disease-indications and management: a review and consensus report by the therapeutic apheresis subsection of the AABB. Transfusion. 2018;58(8):1965-1972. doi:10.1111/trf.14806
17. Han H, Hensch L, Tubman VN. Indications for transfusion in the management of sickle cell disease. Hematology Am Soc Hematol Educ Program. 2021;2021(1):696-703. doi:10.1182/hematology.2021000307
Prognosis for Sickle Cell Disease
The life span of homozygous patients has steadily increased to > 50 years (1, 2). Common causes of death are acute chest syndrome, intercurrent infections, pulmonary emboli, infarction of a vital organ, pulmonary hypertension, and chronic kidney disease.
Prognosis references
1. DeBaun MR, Ghafuri DL, Rodeghier M, et al. Decreased median survival of adults with sickle cell disease after adjusting for left truncation bias: a pooled analysis. Blood. 2019;133(6):615-617. doi:10.1182/blood-2018-10-880575
2. Wierenga KJ, Hambleton IR, Lewis NA. Survival estimates for patients with homozygous sickle-cell disease in Jamaica: a clinic-based population study. Lancet. 2001;357(9257):680-683. doi:10.1016/s0140-6736(00)04132-5
Key Points
Patients homozygous for hemoglobin S (Hb S) have an abnormal beta chain, resulting in fragile, relatively inflexible red blood cells when Hb S polymerizes; red blood cells can adhere to the endothelium and cause vasoconstriction and inflammation, leading to tissue infarction, and are prone to hemolysis, causing anemia.
Patients have various acute exacerbations including painful crisis, sequestration crisis, aplastic crisis, and acute chest syndrome.
Long-term consequences include pulmonary hypertension, chronic kidney disease, stroke, aseptic necrosis, and increased risk of infection.
Diagnose using hemoglobin electrophoresis.
For acute crises, give opioid analgesics for pain, check for worsening anemia (suggesting aplastic or sequestration crisis) and signs of acute chest syndrome or infection, restore normal intravascular volume.
Prevent infection by using vaccinations and prophylactic antibiotics; limit painful crises and risk of disease complications by giving hydroxyurea.Prevent infection by using vaccinations and prophylactic antibiotics; limit painful crises and risk of disease complications by giving hydroxyurea.





