

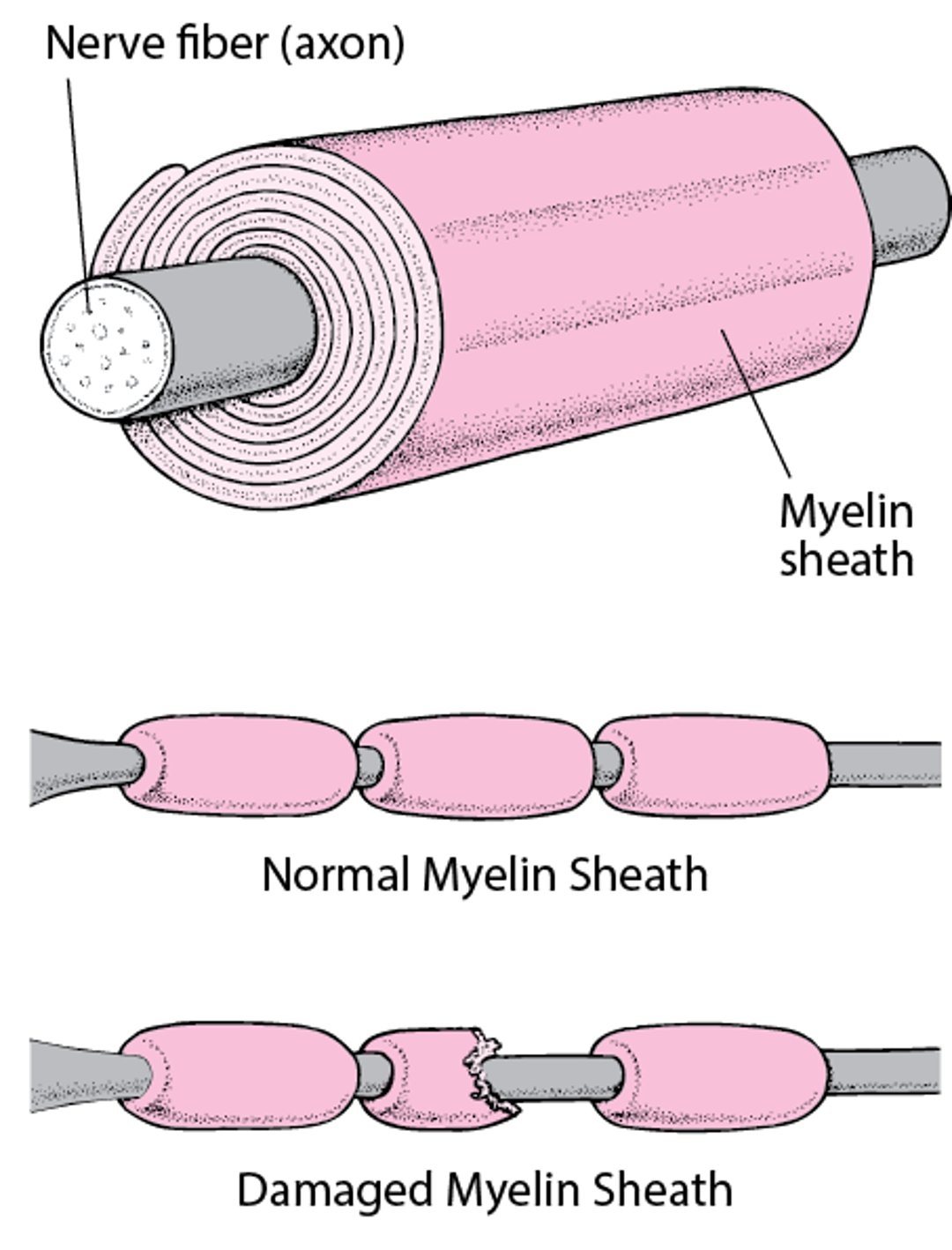
Disorders that cause demyelination and have no known cause are called primary demyelinating disorders. Demyelination is the destruction of the tissues that wrap around nerves, called the myelin sheath.
Insulating a Nerve Fiber
Most nerve fibers inside and outside the brain are wrapped with many layers of tissue composed of a fat (lipoprotein) called myelin. These layers form the myelin sheath. Much like the insulation around an electrical wire, the myelin sheath enables nerve signals (electrical impulses) to be conducted along the nerve fiber with speed and accuracy. When the myelin sheath is damaged (called demyelination), nerves do not conduct electrical impulses normally. |

Sometimes primary demyelinating disorders develop after a viral infection. A likely explanation is that the virus or another substance somehow triggers the immune system to attack the body’s own tissues (autoimmune reaction). The autoimmune reaction results in inflammation, which damages the myelin sheath and the nerve fiber under it.
Multiple sclerosis is the most common primary demyelinating disorder.
Acute Disseminated Encephalomyelitis (ADEM)
This rare type of inflammation leads to demyelination of nerves in the brain and spinal cord. Acute disseminated encephalomyelitis is more common among children than among adults.
Acute disseminated encephalomyelitis usually develops after a viral infection. Acute disseminated encephalomyelitis is thought to be a misguided immune reaction triggered by the virus. In the United States, this disorder usually results from some types of influenza, hepatitis A, hepatitis B, or infection with enteroviruses, Epstein-Barr virus, or human immunodeficiency virus (HIV). Measles, chickenpox, and rubella used to be common causes before childhood vaccination became widespread.
Typically, the inflammation develops 1 to 3 weeks after the viral illness begins.
Symptoms of ADEM
Symptoms of acute disseminated encephalomyelitis appear rapidly. At first, people may have a fever, a headache, nausea, and vomiting and feel tired. When the disorder is severe, it can cause seizures and coma.
Vision in one or both eyes may be lost. Muscles may become weak, and coordination may be impaired, making walking difficult. People may become paralyzed. Sensation may be lost in parts of the body, making them feel numb. Mental function (including thinking, judgment, and learning) may be affected.
Most people recover within days, and within 6 months, most have totally or almost totally recovered. Other people may remain impaired the rest of their life. Muscles may remain weak, and areas of the body may remain numb. People may not recover their vision or mental function.
Diagnosis of ADEM
Medical history and physical examination
Doctors may be able to diagnose acute disseminated encephalomyelitis based on symptoms and results of a physical examination. Magnetic resonance imaging (MRI) may be done.
A spinal tap (lumbar puncture) may be done to check for meningitis or a brain infection (encephalitis). Blood tests may be done to check for other disorders that cause similar symptoms.
Treatment of ADEM
Steroids (sometimes called corticosteroids or glucocorticoids)
Immune globulin or plasma exchange
Acute disseminated encephalomyelitis can be treated with steroids given intravenously.
Immune globulin and plasma exchange may also be effective. These treatments may be used with or without steroids. Immune globulin consists of antibodies obtained from the blood of people with a normal immune system. For plasma exchange, blood is withdrawn, abnormal antibodies are removed from it, and the blood is returned to the person.

Adrenoleukodystrophy and Adrenomyeloneuropathy
Adrenoleukodystrophy and adrenomyeloneuropathy are rare hereditary metabolic disorders. In these disorders, fats are not broken down as they normally are. These fats accumulate mainly in the brain, spinal cord, and adrenal glands. In the brain, they cause demyelination of nerves.
Adrenoleukodystrophy affects young boys, usually between the ages of 4 and 8. A milder, more slowly developing form of the disorder can begin during adolescence or young adulthood.
Adrenomyeloneuropathy is a milder form. It begins when men are in their 20s or 30s.
In these disorders, widespread demyelination is often accompanied by adrenal gland dysfunction. Boys have behavioral problems and problems with hearing and vision. Eventually, mental deterioration, involuntary and uncoordinated muscle contractions (spasticity), and blindness occur. Some boys with adrenoleukodystrophy are totally disabled or die 2 to 3 years after diagnosis. Often, adults with adrenomyeloneuropathy first notice a problem when their legs become weak and stiff, they lose control of their bladder or bowels (incontinence), and/or erectile dysfunction develops.
The diagnosis of adrenoleukodystrophy or adrenomyeloneuropathy is confirmed by genetic testing.
No cure for either disorder is known. Dietary supplements with glycerol trioleate and glycerol trierucate (known as Lorenzo’s oil) may decrease risk of MRI abnormalities in asymptomatic boys, but its role in symptomatic disease is uncertain.
When the adrenal gland (but not the brain) is affected, treatment with adrenal hormones may be lifesaving. Many experts now recommend stem cell transplantation early in the disease, before serious symptoms develop.
Leber Hereditary Optic Neuropathy
Leber hereditary optic neuropathy causes demyelination leading to partial loss of vision.
Leber hereditary optic neuropathy is more common among men. Usually, symptoms begin between the ages of 15 and 35. This disorder is inherited through the mother, and the defective genes seem to be located in mitochondria (structures in cells that provide energy for the cell).
Vision may become blurred in one eye or in both eyes at the same time. But if vision in one eye is affected, vision in the other eye begins to be lost within weeks or months. The sharpness of vision (acuity) and color vision deteriorate over time.
Some people also have heart problems or muscle symptoms (such as involuntary muscle contractions, muscle weakness, or muscle spasms), which may resemble symptoms of multiple sclerosis.
Doctors can often diagnose Leber hereditary optic neuropathy based on symptoms and results of a physical examination. Testing can identify some of the abnormal genes responsible for the disorders. Electrocardiography is done to check for the heart problems.
There is no established treatment for Leber hereditary optic neuropathy. However, some evidence suggests that the medications idebenone (approved in some countries) and ubiquinone may improve sight in people with Leber hereditary optic neuropathy. Gene therapy, involving injecting the normal gene into the eye, has shown promising results when treatment is begun within 12 months of symptoms; further studies are ongoing.
Limiting consumption of alcohol and not using tobacco products may help. Alcohol and tobacco may affect the mitochondria, which is where the defective gene that causes Leber hereditary optic neuropathy is located.





