

Multiple sclerosis (MS) is characterized by disseminated patches of demyelination in the brain and spinal cord. Common symptoms include visual and oculomotor abnormalities, paresthesias, weakness, spasticity, urinary dysfunction, and mild cognitive symptoms. Typically, neurologic deficits are multiple, with remissions and exacerbations gradually producing disability. Diagnosis requires clinical or MRI evidence of ≥ 2 characteristic lesions in the central nervous system that are separated in both time and space (location in the central nervous system). Treatment includes corticosteroids for acute exacerbations, disease-modifying therapies (DMTs) to prevent exacerbations, and supportive measures including rehabilitative care and symptom management.
Multiple sclerosis is believed to involve an immunologic mechanism. One postulated cause is infection by a latent virus (possibly a human herpesvirus such as Epstein-Barr virus), which, when activated, triggers a secondary autoimmune response.
An increased incidence among certain families and presence of human leukocyte antigen (HLA) allotypes (HLA-DR2) suggests genetic susceptibility.
Worldwide, about 2.8 million people have multiple sclerosis (1). The prevalence of MS is higher among people who spend their first 15 years of life in temperate climates (1/2000) than in those who spend them in the tropics (1/10,000) (2). One possible explanation is that lower levels of vitamin D (as found in temperate climates with decreased sun exposure) are associated with an increased risk of MS (3). Cigarette smoking also appears to increase risk.
Age at onset ranges from 15 to 60 years, typically 20 to 40 years; women are affected somewhat more often.
General references
1. Walton C, King R, Rechtman L, et al. Rising prevalence of multiple sclerosis worldwide: Insights from the Atlas of MS, third edition. Mult Scler. 2020;26(14):1816-1821. doi:10.1177/1352458520970841
2. Portaccio E, Magyari M, Havrdova EK, et al. Multiple sclerosis: emerging epidemiological trends and redefining the clinical course. Lancet Reg Health Eur. 2024;44:100977. Published 2024 Aug 22. doi:10.1016/j.lanepe.2024.100977
3. Sintzel MB, Rametta M, Reder AT. Vitamin D and Multiple Sclerosis: A Comprehensive Review. . Vitamin D and Multiple Sclerosis: A Comprehensive Review.Neurol Ther. 2018;7(1):59-85. doi:10.1007/s40120-017-0086-4
Pathophysiology of Multiple Sclerosis
Localized areas of demyelination (plaques) occur, with destruction of oligodendroglia, perivascular inflammation, and chemical changes in lipid and protein constituents of myelin in and around the plaques. Axonal damage is common, and neuronal cell bodies may also die or be damaged.
Astrogliosis develops in plaques that are disseminated throughout the central nervous system (CNS), primarily in white matter, particularly in the lateral and posterior columns (especially in the cervical regions), optic nerves, and periventricular areas. Tracts in the midbrain, pons, and cerebellum are also affected. Gray matter in the cerebrum and spinal cord can also be affected.
Symptoms and Signs of Multiple Sclerosis
Multiple sclerosis is characterized by varied CNS deficits, with remissions and recurring exacerbations. When MS is not treated with immunomodulating medications, exacerbations average about 1 every 2 years, but frequency varies greatly.
Although MS may progress and regress unpredictably, there are typical patterns of progression:
Relapsing-remitting pattern: Exacerbations alternate with remissions, when partial or full recovery occurs or symptoms are stable. Remissions may last months or years. Exacerbations can occur spontaneously or can be triggered by an infection such as influenza. Relapsing forms of MS include active secondary MS (defined as a clinical relapse, a new lesion seen on an MRI scan of the brain or spinal cord, or progressive disability).
Secondary progressive pattern: This pattern begins with relapses alternating with remissions (relapsing-remitting pattern), followed by gradual progression of the disease.
Primary progressive pattern: The disease progresses gradually with no remissions, although there may be temporary plateaus during which the disease does not progress. Unlike in the relapsing-remitting pattern, there are no clear exacerbations.
Progressive relapsing pattern: The disease progresses gradually, but progression is interrupted by sudden, clear relapses. This pattern is rare.
The most common initial symptoms of multiple sclerosis are the following:
Paresthesias in one or more extremities, in the trunk, or on one side of the face
Weakness or clumsiness of a leg or hand
Visual disturbances (eg, partial loss of vision and pain in one eye due to retrobulbar optic neuritis, diplopia due to internuclear ophthalmoplegia, scotomas)
Other common early symptoms of MS include slight stiffness or unusual fatigability of a limb, minor gait disturbances, vertigo, and mild affective disturbances; all usually indicate scattered CNS involvement and may be subtle. Most patients with MS have difficulty with bladder control (eg, frequency, urgency, hesitancy, incontinence, retention). Fatigue is common. Excess heat (eg, warm weather, a hot bath, fever) may temporarily exacerbate symptoms and signs (Uhthoff phenomenon).
Cognitive symptoms are common. Apathy, poor judgment, or inattention may occur. Affective disturbances are common and may include depression (most frequent), emotional lability, or euphoria. Depression may be reactive or partly due to cerebral lesions of MS. A few patients have seizures.
Cranial nerves
Unilateral or asymmetric optic neuritis and bilateral internuclear ophthalmoplegia are typical.
Central vision is affected more than peripheral vision.
Optic neuritis causes loss of vision (ranging from scotomas to blindness), eye pain during eye movement, and sometimes abnormal visual fields, a swollen optic disk, or a partial or complete afferent pupillary defect.
Internuclear ophthalmoplegia results if there is a lesion in the medial longitudinal fasciculus connecting the nuclei of cranial nerves III, IV, and VI. During horizontal gaze, adduction of 1 eye is decreased, with nystagmus of the other (abducting) eye; convergence is intact. Internuclear ophthalmoplegia in MS is typically bilateral; unilateral internuclear ophthalmoplegia is often caused by ischemic stroke.
Rapid, small-amplitude eye oscillations in straight-ahead (primary) gaze (pendular nystagmus) are uncommon but characteristic of MS. Vertigo is common. Intermittent unilateral facial numbness or pain (resembling trigeminal neuralgia), palsy, or spasm may occur. Mild dysarthria may occur, caused by bulbar weakness, cerebellar damage, or disturbance of cortical control. Other cranial nerve deficits are unusual but may occur secondary to brain stem injury.
Motor
Weakness is common and usually reflects corticospinal tract damage in the spinal cord. The weakness preferentially affects the lower extremities and is bilateral and spastic.
Deep tendon reflexes (eg, knee and ankle jerks) are usually increased, and an extensor plantar response (Babinski sign) and clonus are often present. Spastic paraparesis produces a stiff, imbalanced gait; in advanced cases, it may confine patients to a wheelchair. Painful flexor spasms in response to sensory stimuli (eg, bedclothes) may occur. Cerebral or cervical spinal cord lesions may result in hemiparesis, which sometimes is the presenting symptom.
Reduced mobility increases the risk of osteoporosis.
Cerebellar
In advanced MS, cerebellar ataxia plus spasticity may be severely disabling; other cerebellar manifestations include slurred speech, scanning speech (slow enunciation with a tendency to hesitate at the beginning of a word or syllable), and Charcot triad (intention tremor, scanning speech, and nystagmus).
Sensory
Paresthesias and partial loss of any type of sensation are common and often localized (eg, to one or both hands or legs).
Various painful sensory disturbances (eg, burning or electric shocklike pains) can occur spontaneously or in response to touch, especially if the spinal cord is affected. An example is Lhermitte sign, an electric shocklike pain that radiates down the spine or into the legs or arms when the neck is flexed.
Objective sensory changes as demonstrated on neurological examination tend to be transient and difficult to demonstrate early in the disease.
Spinal cord
MS spinal cord involvement most commonly causes bladder dysfunction (eg, urinary urgency or hesitancy, partial retention of urine, mild urinary incontinence). Constipation, erectile dysfunction in men, and genital anesthesia in women may occur. Frank urinary and fecal incontinence may occur in advanced MS.
Spinal cord lesions (plaques) are a common source of neuropathic pain.
Diagnosis of Multiple Sclerosis
Clinical criteria based on history, physical examination, and radiologic findings
Brain and spinal MRI
Sometimes cerebrospinal fluid (CSF) IgG levels and evoked potentials
Multiple sclerosis is suspected in patients with optic neuritis, internuclear ophthalmoplegia, or other symptoms that suggest MS, particularly if deficits are multifocal or intermittent. If MS is suspected, brain MRI and spinal MRI are done.
MRI is the most sensitive imaging test for MS and can exclude other treatable disorders that may mimic MS, such as nondemyelinating lesions at the junction of the spinal cord and medulla (eg, subarachnoid cyst, foramen magnum tumors). Gadolinium contrast enhancement can distinguish actively inflamed from older plaques. Also, higher-field MRI magnets (3 to 7 Tesla) can distinguish perivenular MS plaques from nonspecific white matter lesions.

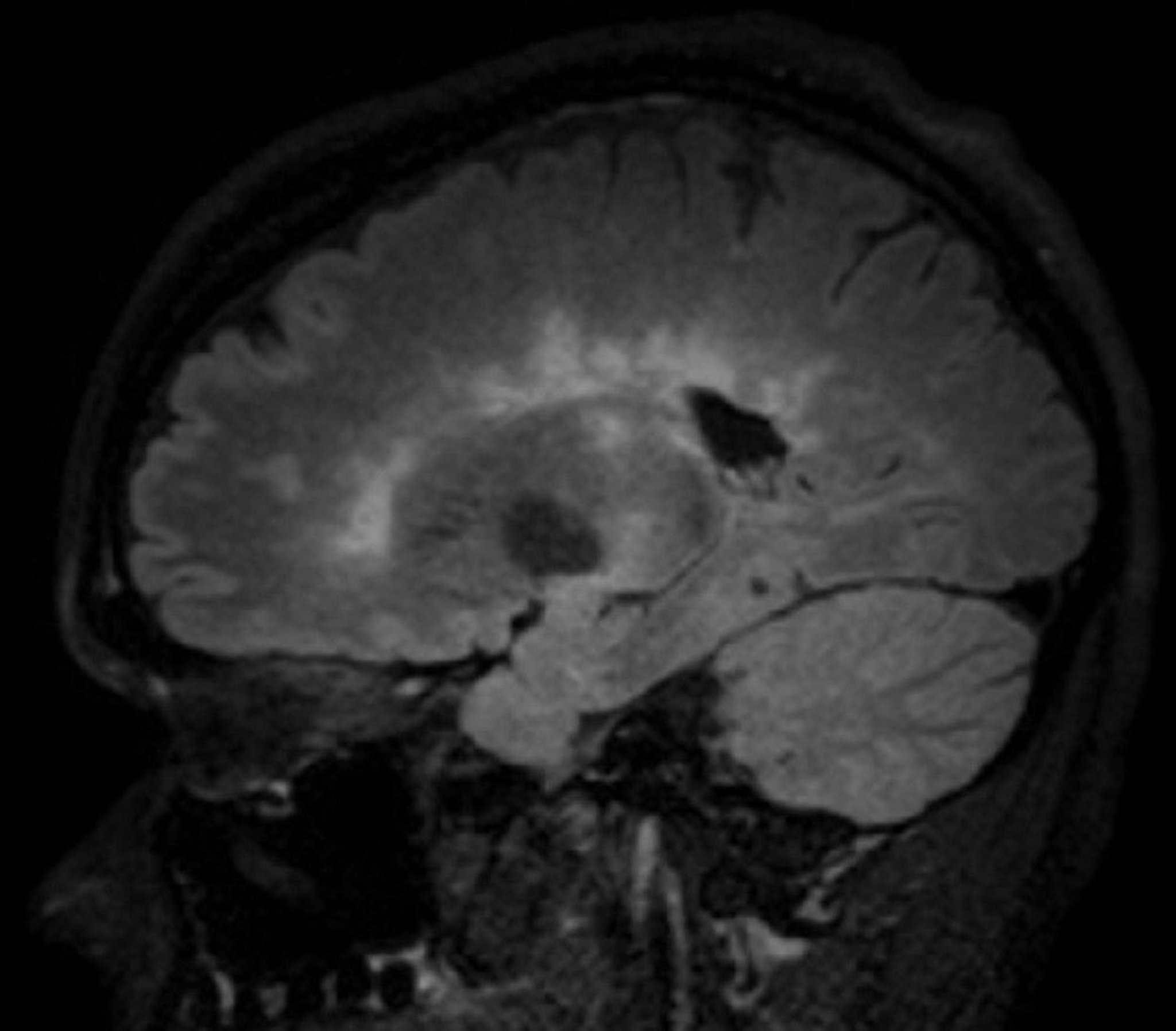
This image shows multiple hyperintense white matter lesions seen in a sagittal view of the brain of a patient with multiple sclerosis. This finding is sometimes called Dawson fingers.
© 2017 Elliot K. Fishman, MD.
MS must be distinguished from the following:
Clinically isolated syndromes (consisting of only a single clinical manifestation typical of MS)
Radiologically isolated syndrome (MRI findings typical of MS that are incidentally noted in patients with no clinical manifestations)
Neuromyelitis optica spectrum disorder (Devic disease), previously considered a variant of MS, now recognized as a separate disorder
The diagnosis of MS requires a clinical episode consistent with MS (such as optic neuritis, partial myelitis or internuclear ophthalmoplegia) plus evidence of CNS lesions that are separated in both time and space (location in the CNS). For example, any of the following can indicate separation in time:
A history of exacerbations and remissions
MRI that shows simultaneous enhancing and nonenhancing lesions, even if patients are asymptomatic
A new lesion on a subsequent MRI in patients with a previous lesion
Separation (dissemination) in space can be established by finding lesions in ≥ 2 of the 5 following CNS areas typically affected by MS (1):
Periventricular: ≥ 1 lesions (2)
Cortical/juxtacortical (white matter next to cortex and/or cortex): ≥ 1 lesions
Infratentorial: ≥ 1 lesions
Spinal cord: ≥ 1 lesions
Optic nerve: ≥ 1 lesions (either by MRI or clinical evaluation)
Additional testing
If MRI plus clinical findings are not diagnostic, additional testing may be necessary to objectively demonstrate separate neurologic abnormalities. Such testing may include evoked potentials and, occasionally, CSF examination or blood tests.
Evoked potentials (delays in electrical responses to sensory stimulation) are often more sensitive for MS than symptoms or signs. Visual evoked responses are sensitive and particularly helpful in patients with no confirmed cranial lesions (eg, those with lesions only in the spinal cord). Somatosensory evoked potentials and brain stem auditory evoked potentials are sometimes also measured.
CSF examination, not routinely performed, can be helpful if MRI plus clinical findings are inconclusive or if infection (eg, CNS Lyme disease) must be ruled out. CSF tests include opening pressure, cell count and differential, protein, glucose, IgG, oligoclonal bands, and usually myelin basic protein and albumin. IgG is usually increased as a percentage of CSF components, such as protein (normally < 11%) or CSF albumin (normally < 27%). IgG levels correlate with disease severity. Oligoclonal IgG bands can usually be detected by electrophoresis of CSF. Myelin basic protein may be elevated during active demyelination. CSF lymphocyte count and protein content may be slightly increased.
Blood tests may be necessary. When clinically indicated by atypical symptoms or geographic area, specific blood testing for inflammatory disorders (eg, SLE) and infections (eg, Lyme disease) should be done to exclude these conditions, which can mimic MS. Blood tests to measure IgG antibodies specific for neuromyelitis optica spectrum disorder (aquaporin-4 antibody [also known as NMO-IgG] and anti-MOG [myelin oligodendrocyte glycoprotein] antibodies) may be done to differentiate that disorder from MS.
Diagnosis references
1. Filippi M, Rocca MA, Ciccarelli O, et al. MRI criteria for the diagnosis of multiple sclerosis: MAGNIMS consensus guidelines. Lancet Neurol. 15 (3):292–303, 2016. doi: 10.1016/S1474-4422(15)00393-2
2. Thompson AJ, Banwell BL, Barkhof F, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018;17:162–173.
Treatment of Multiple Sclerosis
Glucocorticoids for acute exacerbations
Disease-modifying therapies (DMTs) to prevent exacerbations and delay eventual disability
Rehabilitative care and symptom management
Goals for treatment of multiple sclerosis include the following:
Shortening acute exacerbations
Decreasing frequency of exacerbations
Relieving symptoms
Delaying disability, particularly maintaining the patient’s ability to walk
Treatment of exacerbations and relapses
Glucocorticoids, given orally or intravenously in brief courses (3 to 5 days), are used to treat acute onset of symptoms or exacerbations that cause objective deficits sufficient to impair function (eg, loss of vision, strength, or coordination). Glucocorticoids may be given orally (methylprednisolone or prednisone) or intravenously (methylprednisolone). Some data indicate that oral or IV methylprednisolone have similar efficacy (Glucocorticoids, given orally or intravenously in brief courses (3 to 5 days), are used to treat acute onset of symptoms or exacerbations that cause objective deficits sufficient to impair function (eg, loss of vision, strength, or coordination). Glucocorticoids may be given orally (methylprednisolone or prednisone) or intravenously (methylprednisolone). Some data indicate that oral or IV methylprednisolone have similar efficacy (1, 2).
Short courses of high-dose glucocorticoids, oral or intravenous, may shorten acute exacerbations, temporarily slow progression, and improve MRI measures of disease. However, glucocorticoids have not been shown to delay the progression of long-term disability in patients with MS (3).
If glucocorticoids are ineffective in reducing the severity of an exacerbation, plasma exchange may be used (4). Plasma exchange can be used for any relapsing form of MS (relapsing-remitting, progressive relapsing, secondary progressive), but is not used for primary progressive MS.
Plasma exchange and hematopoietic stem cell transplantation (5) may be useful for severe, intractable disease.
Disease-modifying therapies
Disease-modifying therapies (DMTs) target and control the immune system, to prevent exacerbations and delay eventual disability. There are comprehensive practice guidelines for using DMTs (6, 7). For additional information on DMTs, see Practice guideline recommendations summary: Disease-modifying therapies for adults with multiple sclerosis (American Academy of Neurology 2018, reaffirmed 2024), the National Multiple Sclerosis Society website, and updated guidelines on the use of high-efficacy therapies (8–11).
Disease-modifying therapies that treat relapsing remitting MS have been categorized as either moderately efficacious or highly efficacious based on their ability to reduce relapse rate, new MRI lesions, and disability:
Moderately efficacious: interferon beta 1a, interferon beta 1b, glatiramer acetate, teriflunomide, dimethyl fumurate, monomethyl fumurate, diroximel fumarate, fingolimod, siponimod, ozanimod, ponesimodinterferon beta 1a, interferon beta 1b, glatiramer acetate, teriflunomide, dimethyl fumurate, monomethyl fumurate, diroximel fumarate, fingolimod, siponimod, ozanimod, ponesimod
Highly efficacious: natalizumab, rituximab, ocrelizumab, ofatumumab, ublituximab, cladribine, and alemtuzumab. Some experts also include fingolimod, siponimod, ozanimod, ponesimod in this category.natalizumab, rituximab, ocrelizumab, ofatumumab, ublituximab, cladribine, and alemtuzumab. Some experts also include fingolimod, siponimod, ozanimod, ponesimod in this category.
Many of the DMTs are also approved for clinically isolated syndrome and active secondary progressive MS.
The following moderately efficacious disease-modifying therapies are injectable and self-administered:
Interferon beta-1aInterferon beta-1a
Interferon beta-1bInterferon beta-1b
Glatiramer acetateGlatiramer acetate
The following oral immunomodulatory medications, grouped by efficacy, can be used to treat relapsing forms of MS, including active secondary MS (patients transitioning from relapsing remitting MS into secondary progressive MS, but still showing active disease).
Moderate efficacy:
TeriflunomideTeriflunomide
Dimethyl fumarateDimethyl fumarate
Monomethyl fumarateMonomethyl fumarate
Diroximel fumarateDiroximel fumarate
Moderate efficacy (though some consider high efficacy):
FingolimodFingolimod
Siponimod Siponimod
OzanimodOzanimod
PonesimodPonesimod
High efficacy:
CladribineCladribine
There is growing consensus regarding using high-efficacy therapies as first line when initiating treatment. If a patient is already on a disease-modifying therapy, many experts continue this treatment (even if not identified as "high efficacy") until there is evidence of disease worsening, such as a new relapse, new MRI lesion, or worsening disability. Treatment plans, including when to initiate disease-modifying therapy, are best decided with patient education and shared decision making between patients and physicians. If one medication is ineffective, a different one can be tried. Treatments should be tailored to the patient and managed by MS specialists.
Since monoclonal antibodies for MS treatment have become available, the immunosuppressant mitoxantrone is used less often but may still be helpful, particularly for progressive MS that is refractory to other treatments. Since monoclonal antibodies for MS treatment have become available, the immunosuppressant mitoxantrone is used less often but may still be helpful, particularly for progressive MS that is refractory to other treatments.
Natalizumab, an anti–alpha-4 integrin antibody, inhibits passage of leukocytes across the blood-brain barrier; given as a monthly infusion, it reduces number of exacerbations and new brain lesions but may increase the risk of Natalizumab, an anti–alpha-4 integrin antibody, inhibits passage of leukocytes across the blood-brain barrier; given as a monthly infusion, it reduces number of exacerbations and new brain lesions but may increase the risk ofprogressive multifocal leukoencephalopathy (PML). Symptoms of PML include aphasia, change in mental status, hemianopia, and ataxia.
Medications that increase the risk of PML include the following (in descending order of risk):
NatalizumabNatalizumab
FingolimodFingolimod
SiponimodSiponimod
RituximabRituximab
OcrelizumabOcrelizumab
Ofatumumab Ofatumumab
Rarely, dimethyl fumarateRarely, dimethyl fumarate
Consultation with a neurologist with training in MS is highly recommended before using any of these medications. Prior to starting disease-modifying therapies, blood tests should be done to check for antibodies to JC virus (Jakob-Creutzfeldt virus, JCV), which causes PML. Based on the results, one of the following is done:
If results are positive, patients should be counseled about the risk of PML.
If results are negative, antibody tests should be done every 6 months as long as any of these medications is used; seroconversion is common.
If test results become positive, patients should be counseled again about the risk, and clinicians should consider switching to a medication without PML risk.
If the high-risk medication is continued, MRI of the brain should be done about every 6 months.
Development of PML symptoms requires immediate brain MRI, with and without gadolinium. MRI can often distinguish PML from MS. After MRI, a lumbar puncture should be done, and cerebrospinal fluid should be tested for JCV DNA by polymerase chain reaction (PCR). A positive result indicates PML, and emergency consultation with a neurologist and an infectious disease specialist is needed. If patients with a positive result have taken natalizumab, should be done, and cerebrospinal fluid should be tested for JCV DNA by polymerase chain reaction (PCR). A positive result indicates PML, and emergency consultation with a neurologist and an infectious disease specialist is needed. If patients with a positive result have taken natalizumab,plasma exchange can be done to remove the medication quickly, and if immune reconstitution inflammatory syndrome (IRIS) develops, glucocorticoids are given.
Pearls & Pitfalls
|
Alemtuzumab, an anti-CD52 humanized monoclonal antibody given IV, is effective in the treatment of MS but it increases risk of autoimmune disorders, serious infusion reactions, and certain cancers (Alemtuzumab, an anti-CD52 humanized monoclonal antibody given IV, is effective in the treatment of MS but it increases risk of autoimmune disorders, serious infusion reactions, and certain cancers (12). Alemtuzumab is usually used only when treatment with ≥ 2 other medications has been ineffective.). Alemtuzumab is usually used only when treatment with ≥ 2 other medications has been ineffective.
Cladribine may be an appropriate treatment for relapsing MS that is highly active. Cladribine is given orally in 2 yearly treatment courses. Lymphocyte counts should be monitored before, during, and after treatment, and patients should be closely monitored for adverse effects related to immunosupression.Cladribine may be an appropriate treatment for relapsing MS that is highly active. Cladribine is given orally in 2 yearly treatment courses. Lymphocyte counts should be monitored before, during, and after treatment, and patients should be closely monitored for adverse effects related to immunosupression.
Ocrelizumab, an anti-CD20 (B-cell) humanized monoclonal antibody given as an infusion every 6 months, is also effective in the treatment of relapsing MS (Ocrelizumab, an anti-CD20 (B-cell) humanized monoclonal antibody given as an infusion every 6 months, is also effective in the treatment of relapsing MS (13). Ocrelizumab can also be used to treat primary progressive MS, typically in ambulatory patients.). Ocrelizumab can also be used to treat primary progressive MS, typically in ambulatory patients.
Ofatumumab, also an anti-CD20 (B-cell) humanized monoclonal antibody, is used to treat relapsing forms of MS, including clinically isolated syndrome and active secondary progressive disease. It is given by subcutaneous injection (self-administered [Ofatumumab, also an anti-CD20 (B-cell) humanized monoclonal antibody, is used to treat relapsing forms of MS, including clinically isolated syndrome and active secondary progressive disease. It is given by subcutaneous injection (self-administered [14]).
Ublituximab, an anti-CD20 (B-cell) chimeric monoclonal antibody is also used to treat relapsing forms of MS, including clinically isolated syndrome, and active secondary progressive disease. It is given as an IV infusion.Ublituximab, an anti-CD20 (B-cell) chimeric monoclonal antibody is also used to treat relapsing forms of MS, including clinically isolated syndrome, and active secondary progressive disease. It is given as an IV infusion.
Rituximab, an anti-CD20 (B-cell) (used off label for MS in the United States) is also more effective than glatiramer and the interferons (Rituximab, an anti-CD20 (B-cell) (used off label for MS in the United States) is also more effective than glatiramer and the interferons (15); it is commonly used throughout Europe and Canada because it is much less expensive than ocrelizumab. ); it is commonly used throughout Europe and Canada because it is much less expensive than ocrelizumab.
Immunosuppressants other than mitoxantrone (eg, methotrexate, azathioprine, mycophenolate, cyclophosphamide, cladribine) have been used for more severe, progressive MS but are controversial.Immunosuppressants other than mitoxantrone (eg, methotrexate, azathioprine, mycophenolate, cyclophosphamide, cladribine) have been used for more severe, progressive MS but are controversial.
Rehabilitative care and symptom management
Patients with multiple sclerosis are helped by encouragement and reassurance from their care providers.
Physical, occupational, and speech therapists should be consulted as needed and be integrated into the care plan of an individual patient. Regular exercise (eg, stationary biking, treadmill, swimming, stretching, balance exercises), with or without physical therapy, is recommended, even for patients with advanced MS. Exercise conditions the heart and muscles, reduces spasticity, prevents contractures and falls, and has psychological benefits.
Vitamin D supplements (eg, 600 to 4000 IU/day to achieve blood levels of 20 to 50 ng/mL [50 to 125 nmol/L]) are generally recommended based on mixed evidence that they may delay the risk of disease progression; however, this effect has not been consistently shown in clinical trials (Vitamin D supplements (eg, 600 to 4000 IU/day to achieve blood levels of 20 to 50 ng/mL [50 to 125 nmol/L]) are generally recommended based on mixed evidence that they may delay the risk of disease progression; however, this effect has not been consistently shown in clinical trials (16–18). Serum vitamin D levels should be monitored to make sure that dosing is adequate. Vitamin D also reduces the risk of osteoporosis, particularly in patients at increased risk because of decreased mobility or glucocorticoid therapy.levels should be monitored to make sure that dosing is adequate. Vitamin D also reduces the risk of osteoporosis, particularly in patients at increased risk because of decreased mobility or glucocorticoid therapy.
Patients should maintain as normal and active a life as possible but should avoid overwork, fatigue, and exposure to excess heat. Cigarette smoking or other nicotine ingestions should be stopped.Patients should maintain as normal and active a life as possible but should avoid overwork, fatigue, and exposure to excess heat. Cigarette smoking or other nicotine ingestions should be stopped.
Vaccination does not appear to increase risk of exacerbations.
Debilitated patients require measures to prevent pressure ulcers and urinary tract infections; intermittent urinary self-catheterization may be necessary.
Symptom control
Other treatments can be used to control specific symptoms:
Spasticity is treated with escalating doses of baclofen or tizanidine. Injectable botulinum toxin in spastic muscles is used if oral therapies are ineffective (is treated with escalating doses of baclofen or tizanidine. Injectable botulinum toxin in spastic muscles is used if oral therapies are ineffective (19). Gait training and range-of-motion exercises can help weak, spastic limbs.
Problems with gait may be treated with extended-release 4-aminopyridine (dalfampridine).may be treated with extended-release 4-aminopyridine (dalfampridine).
Painful paresthesias are usually treated with gabapentin or pregabalin. Alternatives include tricyclic antidepressants (amitriptyline or desipramine), carbamazepine or other antiseizure medications, and opioids.are usually treated with gabapentin or pregabalin. Alternatives include tricyclic antidepressants (amitriptyline or desipramine), carbamazepine or other antiseizure medications, and opioids.
Depression is treated with counseling and antidepressants.
Bladder dysfunction is treated based on its underlying mechanism.
Constipation may be treated with stool softeners or laxatives, taken regularly.
Fatigue can be treated with amantadine, modafinil, armodafinil, or extended-release amphetamine.can be treated with amantadine, modafinil, armodafinil, or extended-release amphetamine.
Tremor: Tremor associated with multiple sclerosis is difficult to treat. Empiric therapy with clonazepam or gabapentin may help. In severe cases, injecting botulinum toxin into symptomatic muscles may help (Tremor associated with multiple sclerosis is difficult to treat. Empiric therapy with clonazepam or gabapentin may help. In severe cases, injecting botulinum toxin into symptomatic muscles may help (20).
Treatment references
1. Le Page E, Veillard D, Laplaud DA, et al. Oral versus intravenous high-dose methylprednisolone for treatment of relapses in patients with multiple sclerosis (COPOUSEP): A randomised, controlled, double-blind, non-inferiority trial. . Oral versus intravenous high-dose methylprednisolone for treatment of relapses in patients with multiple sclerosis (COPOUSEP): A randomised, controlled, double-blind, non-inferiority trial.Lancet. 386 (9997):974–981, 2015. doi: 10.1016/S0140-6736(15)61137-0
2. Burton JM, O'Connor PW, Hohol M, Beyene J. Oral versus intravenous steroids for treatment of relapses in multiple sclerosis. Cochrane Database Syst Rev. 12:CD006921, 2012. doi: 10.1002/14651858.CD006921.pub3
3. Ciccone A, Beretta S, Brusaferri F, Galea I, Protti A, Spreafico C. Corticosteroids for the long-term treatment in multiple sclerosis. Cochrane Database Syst Rev. 2008;(1):CD006264. Published 2008 Jan 23. doi:10.1002/14651858.CD006264.pub2
4. Tumani H. Corticosteroids and plasma exchange in multiple sclerosis. J Neurol. 2008;255 Suppl 6:36-42. doi:10.1007/s00415-008-6007-9
5. Burt RK, Balabanov R, Burman J, et al. Effect of Nonmyeloablative Hematopoietic Stem Cell Transplantation vs Continued Disease-Modifying Therapy on Disease Progression in Patients With Relapsing-Remitting Multiple Sclerosis: A Randomized Clinical Trial. JAMA. 2019;321(2):165-174. doi:10.1001/jama.2018.18743
6. Rae-Grant A, Day GS, Ruth Ann Marrie RA, et al. Practice guideline recommendations summary: Disease-modifying therapies for adults with multiple sclerosis: Report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology. Neurology. 90 (17):777–788, 2018. doi: 10.1212/WNL.0000000000005347. Correction: Neurology. 92(2):112, 2019.
7. National Multiple Sclerosis Society. Disease-Modifying Therapies. Accessed July 7, 2025.
8. Filippi M, Amato MP, Centonze D, et al. Early use of high-efficacy disease-modifying therapies makes the difference in people with multiple sclerosis: an expert opinion. J Neurol. 2022;269:5382-5394.
9. Buron MD, Chalmer TA, Sellebjerg F, et al. Initial high-efficacy disease-modifying therapy in multiple sclerosis: A nationwide cohort study. Neurology. 2020;95(8):e1041-e1051. doi:10.1212/WNL.0000000000010135
10. Freedman MS, Clift F, Devonshire V, et al. First-Line Use of Higher-Efficacy Disease-Modifying Therapies in Multiple Sclerosis: Canadian Consensus Recommendations. Can J Neurol Sci. Published online June 9, 2025. doi:10.1017/cjn.2025.10342
11. Freeman L, Longbrake EE, Coyle PK, et al. High-efficacy therapies for treatment-naive individuals with relapsing-remitting multiple sclerosis. CNS Drugs. 2022;36;1285–1299.
12. Coles AJ, Achiron A, Traboulsee A, et al. Safety and efficacy with alemtuzumab over 13 years in relapsing-remitting multiple sclerosis: final results from the open-label TOPAZ study. . Safety and efficacy with alemtuzumab over 13 years in relapsing-remitting multiple sclerosis: final results from the open-label TOPAZ study.Ther Adv Neurol Disord. 2023;16:17562864231194823. Published 2023 Sep 21. doi:10.1177/17562864231194823
13. Hauser SL, Bar-Or A, Comi G, et al. Ocrelizumab versus interferon beta-1a in relapsing multiple sclerosis. . Ocrelizumab versus interferon beta-1a in relapsing multiple sclerosis.N Engl J Med. 2017;376(3):221-234. doi:10.1056/NEJMoa1601277
14. Hauser SL, Bar-Or A, Cohen JA, et al. Ofatumumab versus teriflunomide in multiple sclerosis. . Ofatumumab versus teriflunomide in multiple sclerosis.N Engl J Med. 2020;383(6):546-557. doi:10.1056/NEJMoa1917246
15. Granqvist M, Boremalm M , Poorghobad A, et al. Comparative effectiveness of rituximab and other initial treatment choices for multiple sclerosis. . Comparative effectiveness of rituximab and other initial treatment choices for multiple sclerosis.JAMA Neurol. 2018;75(3):320-327. doi:10.1001/jamaneurol.2017.4011
16. Thouvenot E, Laplaud D, Lebrun-Frenay C, et al. High-Dose Vitamin D in Clinically Isolated Syndrome Typical of Multiple Sclerosis: The D-Lay MS Randomized Clinical Trial. . High-Dose Vitamin D in Clinically Isolated Syndrome Typical of Multiple Sclerosis: The D-Lay MS Randomized Clinical Trial.JAMA. 2025;333(16):1413-1422. doi:10.1001/jama.2025.1604
17. Jagannath VA, Filippini G, Di Pietrantonj C, et al. Vitamin D for the management of multiple sclerosis. . Vitamin D for the management of multiple sclerosis.Cochrane Database Syst Rev. 2018;9(9):CD008422. Published 2018 Sep 24. doi:10.1002/14651858.CD008422.pub3
18. Feige J, Moser T, Bieler L, Schwenker K, Hauer L, Sellner J. Vitamin D Supplementation in Multiple Sclerosis: A Critical Analysis of Potentials and Threats. . Vitamin D Supplementation in Multiple Sclerosis: A Critical Analysis of Potentials and Threats.Nutrients. 2020;12(3):783. Published 2020 Mar 16. doi:10.3390/nu12030783
19. Safarpour Y, Mousavi T, Jabbari B. Botulinum Toxin Treatment in Multiple Sclerosis-a Review. Curr Treat Options Neurol. 2017;19(10):33. Published 2017 Aug 17. doi:10.1007/s11940-017-0470-5
20. Makhoul K, Ahdab R, Riachi N, et al. Tremor in multiple sclerosis-An overview and future perspectives. Brain Sci. 2020;10(10):722. Published 2020 Oct 12. doi:10.3390/brainsci10100722
Prognosis for Multiple Sclerosis
The course of multiple sclerosis is highly varied and unpredictable. In most patients, especially when MS begins with optic neuritis, remissions can last months to > 10 years.
Most patients (60 to 80% [1]) who initially have a clinically isolated syndrome eventually develop MS, with a second lesion leading to symptoms or detectable by MRI, usually within 5 years after the initial symptoms begin. Treatment with disease-modifying therapies can delay this progression. If patients have a radiologically isolated syndrome without a history of a clinical episode consistent with demyelination, the risk of developing MS is 19 to 90%, depending on the patient's age and the presence of spinal cord or gadolinium-enhancing lesions (2).
Risk for earlier disability is higher if the initial brain or spinal MRI shows more extensive disease, or if patients have motor, bowel, and/or bladder symptoms when they present, or incomplete recovery during relapses. Rapid incapacitation may occur in some patients, such as men with onset in middle age and with frequent exacerbations. Cigarette smoking may accelerate disease progression.
Lifespan is shortened only in very severe cases.
Prognosis references
1. National Multiple Sclerosis Society. Clinically isolated syndrome (CIS). Accessed June 29, 2025.
2. Lebrun-Frénay C, Rollot F, Mondot L, et al. Risk factors and time to clinical symptoms of multiple sclerosis among patients with radiologically isolated syndrome. JAMA Netw Open. 4(10):e2128271, 2021. doi: 10.1001/jamanetworkopen.2021.28271
Key Points
Multiple sclerosis involves demyelination of the CNS; MS may progress unpredictably but has several typical patterns of progression.
The most common symptoms are paresthesias, weakness or clumsiness, and visual symptoms, but a wide variety of symptoms are possible.
MS is confirmed if MRI and clinical findings establish characteristic lesions that are separate in time and space; however, progression to MS is likely if patients have even a single characteristic clinical deficit or possibly a single radiologic lesion.
Treat patients with glucocorticoids (for severe exacerbations) and immunomodulatory medications (to delay or prevent exacerbations).
Treat patients supportively, using rehabilitation services and medications to treat symptoms (eg, spasticity, painful paresthesias, depression, bladder dysfunction, fatigue, gait problems) when warranted.
Drug Information for the Topic





