

Autoimmune hemolytic anemia is caused by autoantibodies that react with red blood cells at temperatures ≥ 37° C (warm antibody hemolytic anemia) or < 37° C (cold agglutinin disease). Hemolysis is predominantly extravascular. The direct antiglobulin (direct Coombs) test establishes the diagnosis and may suggest the cause. Treatment depends on the cause and may include glucocorticoids, splenectomy, IV immune globulin, immunosuppressants, avoidance of triggers (eg, cold), and withdrawal of medications.
Etiology of Autoimmune Hemolytic Anemia
Autoimmune hemolytic anemia is caused by abnormalities extrinsic to the red blood cell (RBC).
Warm antibody hemolytic anemia
Warm antibody hemolytic anemia is the most common form of autoimmune hemolytic anemia (AIHA); it is slightly more common among females (1). Autoantibodies in warm antibody hemolytic anemia react at temperatures ≥ 37° C. Autoimmune hemolytic anemia may be classified as:
Primary (idiopathic)
Secondary (occurring in association with an underlying disorder such as systemic lupus erythematosus (SLE), lymphoma, or chronic lymphocytic leukemia or after use of certain medications)
Some medications (eg, methyldopa, levodopa—see table Some medications (eg, methyldopa, levodopa—see table) stimulate production of autoantibodies against Rh antigens (methyldopa-type of autoimmune hemolytic anemia). Other medications stimulate production of autoantibodies against an antibiotic–RBC-membrane complex as part of a transient hapten mechanism; the hapten may be stable (eg, high-dose penicillin, cephalosporins) or unstable (eg, quinidine, sulfonamides).
In warm antibody hemolytic anemia, hemolysis occurs primarily in the spleen and is not due to direct lysis of RBCs. It may be severe and can be fatal. Most of the autoantibodies in warm antibody hemolytic anemia are IgG. Most are polyclonal panagglutinins against common RBC antigens.
Cold agglutinin disease
Cold agglutinin disease (cold antibody disease) is caused by autoantibodies that react at temperatures < 37° C, but some autoantibodies have a higher thermal amplitude (eg, > 30° C most likely to cause clinical manifestations). Antibody thermal amplitude is more important than its titer; the higher the temperature (ie, the closer to normal body temperature) at which these antibodies react with the RBC, the greater the hemolysis.
Causes include:
Primary or idiopathic (usually associated with a monoclonal B-cell population in individuals considered to have a low-grade lymphoproliferative disorder; antibodies are usually directed against the I antigen)
Secondary cold agglutinin syndrome (occur in the setting of an underlying disorder such as infections, especially mycoplasmal pneumonias or infectious mononucleosis with antibodies directed against the I [mycoplasma] or i [Epstein Barr virus] antigens or an overt lymphoma)
Infections tend to cause acute disease, whereas idiopathic disease (the common form in older adults) tends to be chronic. The chronic hemolysis occurs largely in the extravascular mononuclear phagocyte system of the liver and spleen, but intravascular hemolysis can occur when a complement-amplifying condition, such as infection, is present. The anemia is usually non-severe. Autoantibodies in cold agglutinin disease are almost always clonal IgM.
Paroxysmal cold hemoglobinuria
Paroxysmal cold hemoglobinuria (PCH; Donath-Landsteiner syndrome) is a rare type of cold reactive autoimmune hemolytic anemia. PCH is more common in children. Hemolysis results from exposure to cold, which may even be localized (eg, from drinking cold water, from washing hands in cold water). An IgG antibody binds to the P antigen on RBCs at low temperatures and causes intravascular hemolysis and hemoglobinuria after warming. It occurs most often after a nonspecific viral illness or in otherwise healthy patients, although it occurs in some patients with congenital or acquired syphilis. The severity and rapidity of development of the anemia varies and may be fulminant. In children, this disease is often self-resolving.
Medications That Can Cause Warm Antibody Hemolytic Anemia
Mechanism | Medications |
|---|---|
Autoantibody to Rh antigens | Cephalosporins DiclofenacDiclofenac IbuprofenIbuprofen Interferon alfa LevodopaLevodopa Mefenamic acidMefenamic acid MethyldopaMethyldopa ProcainamideProcainamide Teniposide ThioridazineThioridazine TolmetinTolmetin |
Stable hapten | Cephalosporins Fluorescein sodiumFluorescein sodium Penicillins TetracyclineTetracycline Tolbutamide (not available in the United States) |
Unstable hapten or unknown mechanism | Aminosalicylic acid Amphotericin BAmphotericin B Antazoline Cephalosporins Chlorpropamide DoxepinDoxepin HydrochlorothiazideHydrochlorothiazide IsoniazidIsoniazid ProbenecidProbenecid QuinidineQuinidine QuinineQuinine RifampinRifampin Sulfonamides Thiopental |
Etiology reference
1. Brodsky RA. Warm Autoimmune Hemolytic Anemia. N Engl J Med. 2019;381(7):647-654. doi:10.1056/NEJMcp1900554
Symptoms and Signs of Autoimmune Hemolytic Anemia
Symptoms of warm antibody hemolytic anemia tend to be due to the anemia. If the disorder is severe, fever, chest pain, syncope, or liver or heart failure may occur. Mild splenomegaly is typical. Venous thromboembolic events are common in patients with warm autoimmune hemolytic anemia (1).
Cold agglutinin disease manifests as an acute or chronic hemolytic anemia. Other symptoms or signs include acrocyanosis, Raynaud phenomenon, cold-associated occlusive changes. Cold agglutinin disease is also associated with elevated thrombotic risk (2).
Symptoms of PCH may include severe pain in the back and legs, headache, vomiting, diarrhea, and passage of dark brown urine; hepatosplenomegaly may be present.
Symptoms and signs references
1. Audia S, Bach B, Samson M, et al. Venous thromboembolic events during warm autoimmune hemolytic anemia. PLoS One. 2018;13(11):e0207218. doi:10.1371/journal.pone.0207218
2. Broome CM, Cunningham JM, Mullins M, et al. Increased risk of thrombotic events in cold agglutinin disease: A 10-year retrospective analysis. Res Pract Thromb Haemost. 2020;4(4):628-635. doi:10.1002/rth2.12333
Diagnosis of Autoimmune Hemolytic Anemia
Peripheral smear, reticulocyte count, lactate dehydrogenase (LDH), haptoglobin, indirect bilirubin
Direct antiglobulin test
Cold agglutinin titer and thermal amplitude testing in cold agglutinin disease
Autoimmune hemolytic anemia should be suspected in any patient with a hemolytic anemia (as suggested by the presence of anemia and reticulocytosis). In warm autoimmune hemolytic anemia, the peripheral smear usually shows microspherocytes and a high reticulocyte count with few or no schistocytes, indicating extravascular hemolysis. Laboratory tests typically indicate hemolysis (eg, elevated LDH and indirect bilirubin and decreased haptoglobin). A high mean corpuscular volume (MCV) may occur due to extreme reticulocytosis or agglutination in cold agglutinin disease. Hemolytic anemia in the setting of a low reticulocyte count is rare but can occur due to factors such as renal insufficiency, infection, or bone marrow failure and constitutes a medical emergency requiring prompt transfusions.

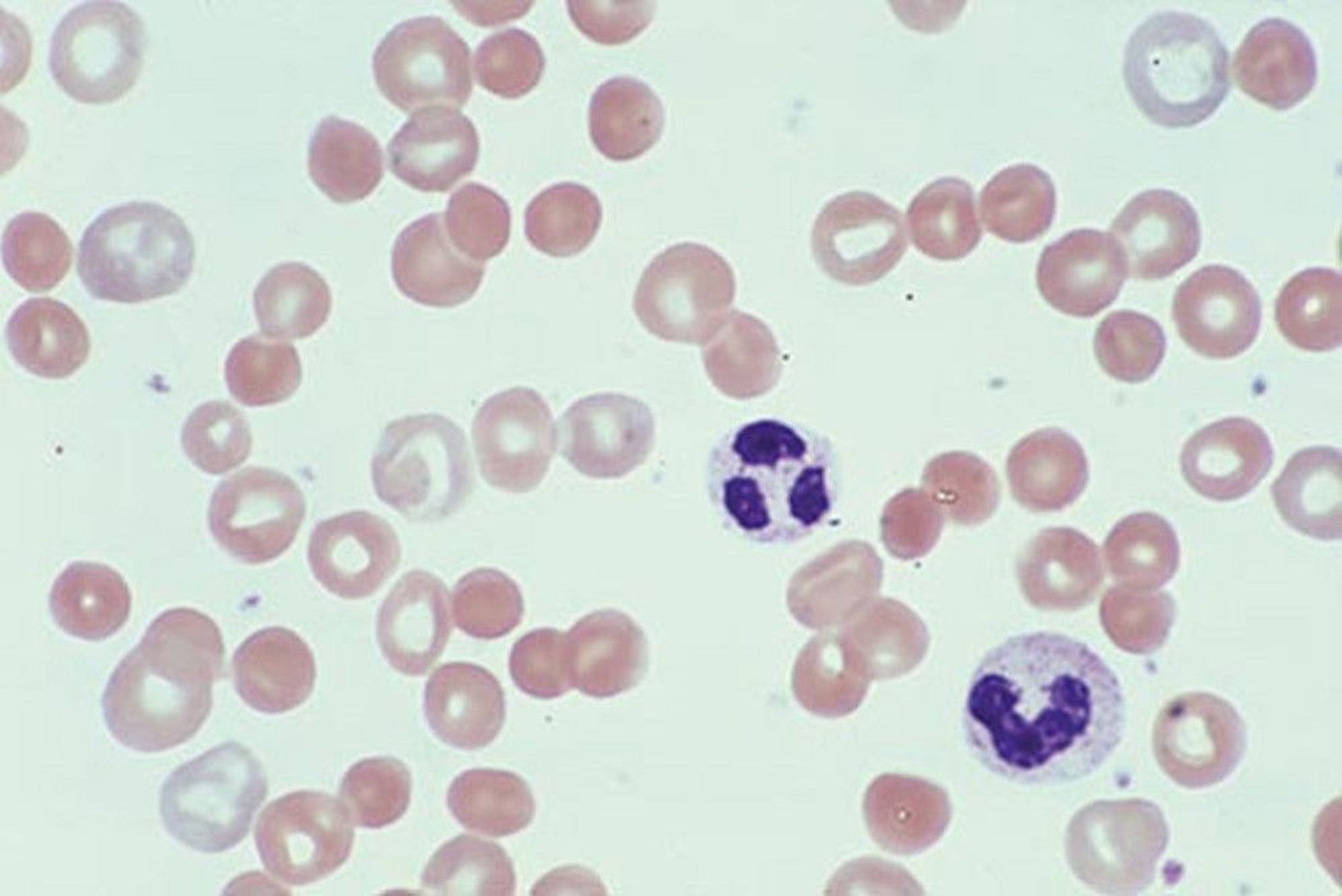
The presence of abundant spherocytes in the peripheral blood smear suggests either hereditary spherocytosis (HS) or autoimmune hemolytic anemia (AIHA). Spherocytes are spherical red blood cells without an area of central pallor and are usually slightly smaller in size than the average red cell. Spherocytes have increased osmotic fragility (because of decreased distensibility associated with reduced surface membrane area) in hypotonic saline, and this test (osmotic fragility test) is positive in HS and in AIHA. The direct antiglobulin (Coombs) test distinguishes between them; the result is positive in AIHA and negative in HS.
By permission of the publisher. From Tefferi A, Li C. In Atlas of Clinical Hematology. Edited by JO Armitage. Philadelphia, Current Medicine, 2004.
Autoimmune hemolytic anemia is diagnosed with the direct antiglobulin (direct Coombs) test (see figure ). Antiglobulin serum is added to washed RBCs from the patient; agglutination indicates the presence of immunoglobulin or complement (C) bound to the RBCs. In warm antibody hemolytic anemia, IgG is nearly always present, and C3 (C3b and C3d) may be present as well. In cold antibody disease, C3 is present while IgG is usually absent. The test is highly sensitive for autoimmune hemolytic anemia with an estimated ~5% of AIHA cases being direct antiglobulin test-negative (1); false-negative results can occur if antibody density is very low or, rarely, if the autoantibodies are IgA.
In most cases of warm antibody hemolytic anemia, the antibody is an IgG identified only as a panagglutinin, meaning the antigen specificity of the antibody cannot be determined. In cold antibody disease, the antibody is usually an IgM directed against the I/i carbohydrate on the RBC surface. Antibody titers can usually be determined but do not always correlate with disease activity. The direct antiglobulin (direct Coombs) test may be positive in the absence of autoimmune hemolytic anemia, and thus should be ordered only in the proper clinical setting. A false-positive direct antiglobulin test may result from the presence of clinically insignificant antibodies or elevated paraproteins, due to IV immune globulin, RhD immunoglobulin, or daratumumab therapy (In most cases of warm antibody hemolytic anemia, the antibody is an IgG identified only as a panagglutinin, meaning the antigen specificity of the antibody cannot be determined. In cold antibody disease, the antibody is usually an IgM directed against the I/i carbohydrate on the RBC surface. Antibody titers can usually be determined but do not always correlate with disease activity. The direct antiglobulin (direct Coombs) test may be positive in the absence of autoimmune hemolytic anemia, and thus should be ordered only in the proper clinical setting. A false-positive direct antiglobulin test may result from the presence of clinically insignificant antibodies or elevated paraproteins, due to IV immune globulin, RhD immunoglobulin, or daratumumab therapy (2). The direct antiglobulin test may also be positive due to alloantibodies after recent transfusion and a delayed hemolytic transfusion reaction.
The indirect antiglobulin (indirect Coombs) test is a complementary test that consists of mixing the patient’s serum or plasma with normal RBCs to determine whether such antibodies are free in the serum or plasma (see figure ). A positive result on an indirect antiglobulin test and a negative result on a direct test generally indicate an alloantibody caused by pregnancy, prior transfusions, or lectin cross-reactivity rather than immune hemolysis. Even identification of a warm antibody does not define hemolysis, because approximately 1/10,000 healthy blood donors has a positive test result (3).
Once autoimmune hemolytic anemia has been identified by the antiglobulin test, testing should differentiate between warm antibody hemolytic anemia and cold agglutinin disease as well as the mechanism responsible for warm antibody hemolytic anemia. This determination can often be made by observing the pattern of the direct antiglobulin reaction. Three patterns are possible:
The reaction is positive with anti-IgG and negative with anti-C3. This pattern is common in idiopathic AIHA and in the drug-associated or methyldopa-type of AIHA, usually warm antibody hemolytic anemia.The reaction is positive with anti-IgG and negative with anti-C3. This pattern is common in idiopathic AIHA and in the drug-associated or methyldopa-type of AIHA, usually warm antibody hemolytic anemia.
The reaction is positive with anti-IgG and anti-C3. This pattern is common in patients with SLE and idiopathic AIHA, usually warm antibody hemolytic anemia, and is rare in drug-associated cases.
The reaction is positive with anti-C3 but negative with anti-IgG. This pattern occurs in cold agglutinin disease (where the antibody is most commonly an IgM). It can also occur in warm antibody hemolytic anemia when the IgG antibody is of low affinity, in some drug-associated cases, and in PCH.
Other tests can suggest the cause of AIHA but are not definitive. For example, in cold agglutinin disease, when unwarmed blood is used, RBCs clump on the peripheral smear, and automated cell counts often reveal an increased mean corpuscular volume and spuriously low hemoglobin due to such clumping; hand warming of the tube and recounting results in values significantly closer to normal.
Thermal amplitude testing in cold agglutinin disease measures the temperature range in which a cold autoantibody binds to its antigen. Cold antibodies that can bind to antigen above 28 to 30° C are considered potentially clinically significant and the closer to core body temperature, the greater the chance that the antibody will cause symptoms and more significant hemolysis.
The cold agglutinin titer is determined by the highest dilution of patient serum able to agglutinate red blood cells at 4°C. A titer of 1:64 is typically part of the diagnosis, though usually titers are higher and occasionally the disease is diagnosed at lower titers.

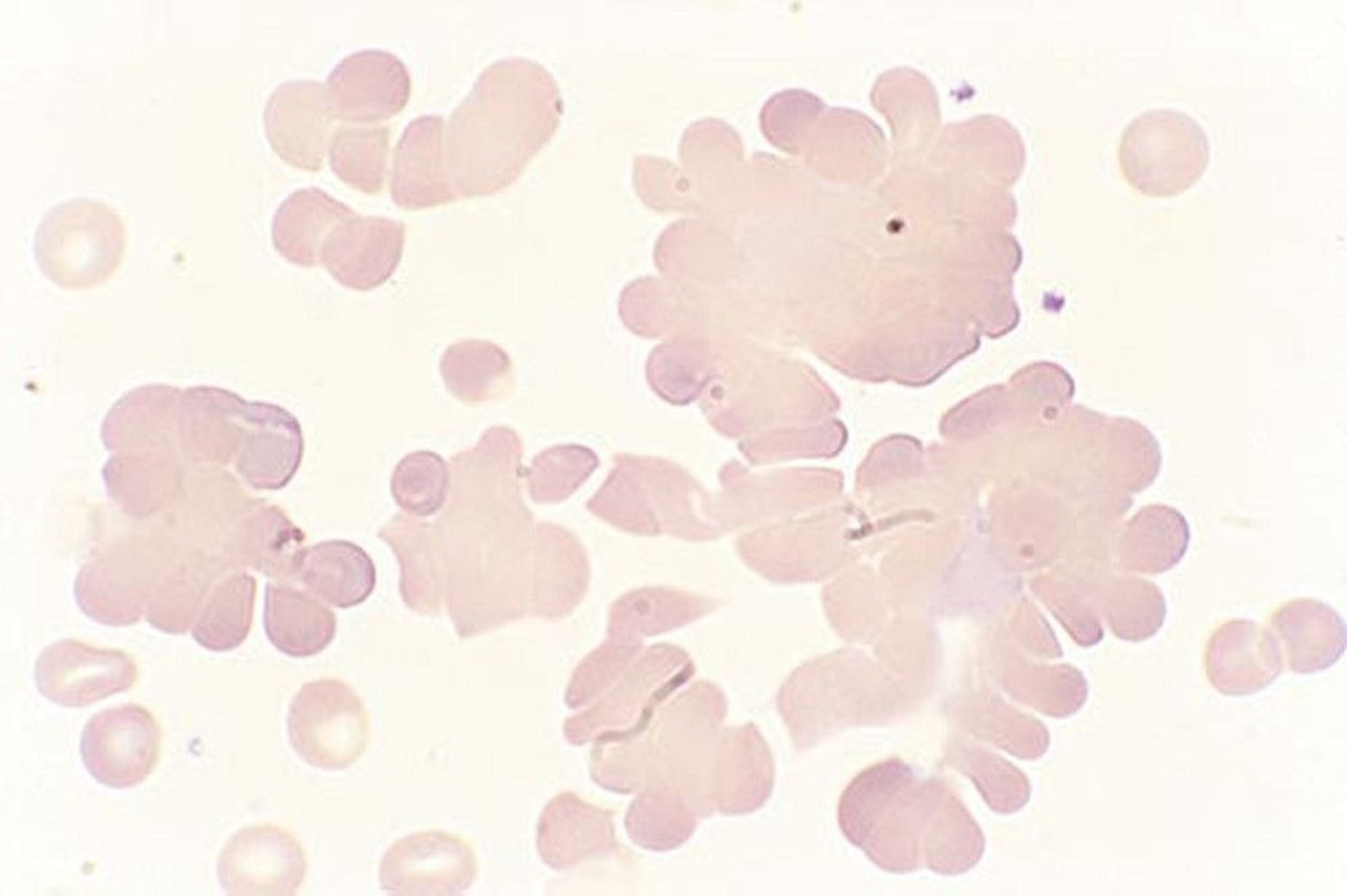
By permission of the publisher. From Tefferi A, Li C. In Atlas of Clinical Hematology. Edited by JO Armitage. Philadelphia, Current Medicine, 2004.
If paroxysmal cold hemoglobinuria (PCH) is suspected, the Donath-Landsteiner test, which is specific for PCH, should be done (4). In this test, the patient's serum is incubated with normal RBCs at 4° C for 30 minutes to allow for fixation of complement and then warmed to body temperature. Hemolysis of the RBCs during this test is indicative of PCH. Because the PCH antibody fixes complement at low temperatures, the direct antiglobulin (direct Coombs) test is positive for C3 and negative for IgG. However, the antibody in PCH is an IgG against the P antigen.
Diagnosis references
1. Sachs UJ, Röder L, Santoso S, Bein G. Does a negative direct antiglobulin test exclude warm autoimmune haemolytic anaemia? A prospective study of 504 cases. Br J Haematol. 2006;132(5):655-656. doi:10.1111/j.1365-2141.2005.05955.x
2. Murphy MF, Dumont LJ, Greinacher A; BEST Collaborative. Interference of New Drugs with Compatibility Testing for Blood Transfusion. N Engl J Med. 2016;375(3):295-296. doi:10.1056/NEJMc1515969
3. Hannon JL. Management of blood donors and blood donations from individuals found to have a positive direct antiglobulin test. Transfus Med Rev. 2012;26(2):142-152. doi:10.1016/j.tmrv.2011.08.004
4. Tiwari AK, Aggarwal G, Mitra S, et al. Applying Donath-Landsteiner test for the diagnosis of paroxysmal cold hemoglobinuria. Asian J Transfus Sci. 2020;14(1):57-59. doi:10.4103/ajts.AJTS_132_17
Treatment of Autoimmune Hemolytic Anemia
Blood transfusion for severe anemia (usually with inappropriate reticulocyte response)
For drug-induced warm antibody hemolytic anemia, drug withdrawal and sometimes IV immune globulin (IVIG)
For idiopathic warm antibody hemolytic anemia, glucocorticoids and, in refractory cases, rituximab, IVIG, immunosuppression (eg, with azathioprine, mycophenolate mofetil, or cyclophosphamide), or splenectomyFor idiopathic warm antibody hemolytic anemia, glucocorticoids and, in refractory cases, rituximab, IVIG, immunosuppression (eg, with azathioprine, mycophenolate mofetil, or cyclophosphamide), or splenectomy
For cold agglutinin disease, avoidance of cold and treatment of underlying disorder; sometimes rituximab with or without chemotherapy, or complement inhibitors For cold agglutinin disease, avoidance of cold and treatment of underlying disorder; sometimes rituximab with or without chemotherapy, or complement inhibitors
For PCH, avoidance of cold, immunosuppressants, and treatment of syphilis if present. In children, this disease is often self-resolving.
Blood transfusion is the most important treatment for patients who have symptoms and who rapidly develop severe, life-threatening anemia. In this situation, transfusion should never be withheld due to lack of "compatible" units. In general, patients who have not had a previous blood transfusion or been pregnant are at low risk for hemolysis of ABO-compatible blood. Even if transfused cells are hemolyzed, blood transfusion can be life-saving until more definitive therapy can be done. Erythropoietin may be given if the reticulocyte response is inadequate.
More specific treatment depends on the mechanism of the hemolysis.
Warm antibody hemolytic anemias
In drug-induced warm antibody hemolytic anemias, withdrawal of the medication decreases the rate of hemolysis. With methyldopa-type AIHA, hemolysis usually ceases within 3 weeks; however, a positive antiglobulin test may persist for withdrawal of the medication decreases the rate of hemolysis. With methyldopa-type AIHA, hemolysis usually ceases within 3 weeks; however, a positive antiglobulin test may persist for> 1 year. With hapten-mediated AIHA, hemolysis ceases when the medication is cleared from the plasma. Glucocorticoids and/or infusions of immune globulin may be used as second-line therapies (1, 2).
In idiopathic warm antibody AIHA, glucocorticoids (eg, prednisone 1 mg/kg orally once a day) are the standard first-line treatment. When stable RBC values are achieved, the glucocorticoids are tapered slowly with laboratory monitoring of hemolysis (eg, by hemoglobin and reticulocyte counts). The goal is to wean the patient completely from glucocorticoids or to maintain remission with the lowest possible glucocorticoid dose. Approximately two-thirds to 80% of patients respond to initial glucocorticoid treatment, however relapse or glucocorticoid dependence is common (glucocorticoids (eg, prednisone 1 mg/kg orally once a day) are the standard first-line treatment. When stable RBC values are achieved, the glucocorticoids are tapered slowly with laboratory monitoring of hemolysis (eg, by hemoglobin and reticulocyte counts). The goal is to wean the patient completely from glucocorticoids or to maintain remission with the lowest possible glucocorticoid dose. Approximately two-thirds to 80% of patients respond to initial glucocorticoid treatment, however relapse or glucocorticoid dependence is common (3, 4, 5). In patients who relapse after glucocorticoid cessation or who are refractory to glucocorticoids, rituximab is usually used as a second-line drug. Many experts also add rituximab to first-line treatment, especially in severe cases (). In patients who relapse after glucocorticoid cessation or who are refractory to glucocorticoids, rituximab is usually used as a second-line drug. Many experts also add rituximab to first-line treatment, especially in severe cases (6). Three dosing regimens of rituximab are used in clinical practice. The standard dose is 375 mg/m² IV weekly for 4 weeks; this is the most commonly used and best-studied regimen. Alternatives include a fixed high dose of 1000 mg IV on day 1 and day 15 (2 doses, 2 weeks apart) , which has been used in some randomized trials, and a low fixed dose of 100 mg IV weekly for 4 weeks. Both fixed doses appear equally efficacious in warm AIHA but not in cold agglutinin disease; however, comparative trials have not been reported. ). Three dosing regimens of rituximab are used in clinical practice. The standard dose is 375 mg/m² IV weekly for 4 weeks; this is the most commonly used and best-studied regimen. Alternatives include a fixed high dose of 1000 mg IV on day 1 and day 15 (2 doses, 2 weeks apart) , which has been used in some randomized trials, and a low fixed dose of 100 mg IV weekly for 4 weeks. Both fixed doses appear equally efficacious in warm AIHA but not in cold agglutinin disease; however, comparative trials have not been reported.
Other treatments include use of additional immunosuppressants, folic acid, and/or splenectomy. Approximately one-third to one-half of patients have a sustained response after splenectomy (Other treatments include use of additional immunosuppressants, folic acid, and/or splenectomy. Approximately one-third to one-half of patients have a sustained response after splenectomy (7).
In cases of fulminant hemolysis, immunosuppression with high-dose pulse glucocorticoids or cyclophosphamide can be used. For less severe but uncontrolled hemolysis, IVIG infusions have provided temporary control. In cases of fulminant hemolysis, immunosuppression with high-dose pulse glucocorticoids or cyclophosphamide can be used. For less severe but uncontrolled hemolysis, IVIG infusions have provided temporary control.
Long-term management with immunosuppressants (including cyclophosphamide, mycophenolate mofetil, azathioprine, sirolimus, cyclosporine, and fostamatinib) has been effective in patients in whom glucocorticoids and splenectomy have been ineffective (Long-term management with immunosuppressants (including cyclophosphamide, mycophenolate mofetil, azathioprine, sirolimus, cyclosporine, and fostamatinib) has been effective in patients in whom glucocorticoids and splenectomy have been ineffective (6, 8).
The presence of panagglutinating antibodies in warm antibody hemolytic anemia makes cross-matching of donor blood difficult. In addition, transfusions could superimpose an alloantibody on the autoantibody, accelerating hemolysis. Thus, transfusions should be administered judiciously when anemia is not severe but should not be withheld in patients with severe or progressive autoimmune hemolytic anemia, particularly when the reticulocyte count is insufficient to compensate.
Because rates of venous thromboembolism are increased in patients with warm AIHA, patients should be maintained on pharmacologic prophylaxis while hospitalized (6, 9).
Cold agglutinin disease
In many cases, avoidance of cold environments and other triggers of hemolysis may be all that is needed to prevent symptomatic anemia.
In cases that occur in a patient with a lymphoma, treatment is directed at the underlying disorder. For primary cold agglutinin disease, rituximab, sometimes with bendamustine, is commonly used, and chemotherapy regimens used to treat B-cell lymphoproliferative disorders can be effective. In cases that occur in a patient with a lymphoma, treatment is directed at the underlying disorder. For primary cold agglutinin disease, rituximab, sometimes with bendamustine, is commonly used, and chemotherapy regimens used to treat B-cell lymphoproliferative disorders can be effective.
Complement inhibition with sutimlimab may be used in patients with severe anemia whose disease requires a more rapid response to therapy or in those with symptomatic anemia to allow time for rituximab or other B-cell depleting therapies to take effect. Complement inhibition with sutimlimab may be used in patients with severe anemia whose disease requires a more rapid response to therapy or in those with symptomatic anemia to allow time for rituximab or other B-cell depleting therapies to take effect.Sutimlimab is also used in patients with symptomatic anemia without a response to B-cell depleting therapies or in those with a contraindication or preference to avoid B-cell depleting therapies. In a clinical trial, sutimlimab, an inhibitor of the classical complement pathway, was shown to increase hemoglobin levels and decrease transfusion requirements in approximately half of patients with cold agglutinin disease (10). However, sutimlimab does not improve cold-induced symptoms, such as Raynaud phenomenon or acrocyanosis. , Vaccination against infectious agents, particularly encapsulated bacteria, is recommended at least 2 weeks prior to treatment with sutimlimab, or antimicrobial prophylaxis may be used.
In severe cases, plasmapheresis is an effective temporary treatment. Transfusions should be given for severe anemia, with the blood and fluids warmed through an on-line warmer.
Splenectomy is usually of no value, and glucocorticoids have limited effectiveness and should not be used as a treatment.
Paroxysmal cold hemoglobinuria
In PCH, therapy consists of strict avoidance of exposure to cold. Glucocorticoids have been effective, but use should be restricted to patients with progressive or idiopathic cases and their effects have not been systematically studied in randomized trials.
Splenectomy is of no value.
Treatment of concomitant syphilis may cure PCH.
Treatment references
1. Garratty G. Immune hemolytic anemia associated with drug therapy. Blood Rev. 2010;24(4-5):143-150. doi:10.1016/j.blre.2010.06.004
2. Maquet J, Lafaurie M, Michel M, Lapeyre-Mestre M, Moulis G. Drug-induced immune hemolytic anemia: detection of new signals and risk assessment in a nationwide cohort study. Blood Adv. 2024;8(3):817-826. doi:10.1182/bloodadvances.2023009801
3. Abdallah GEM, Abbas WA, Elbeih EAS, Abdelmenam E, Mohammed Saleh MF. Systemic corticosteroids in the treatment of warm autoimmune hemolytic anemia: A clinical setting perspective. Blood Cells Mol Dis. 2021;92:102621. doi:10.1016/j.bcmd.2021.102621
4. Birgens H, Frederiksen H, Hasselbalch HC, et al. A phase III randomized trial comparing glucocorticoid monotherapy versus glucocorticoid and rituximab in patients with autoimmune haemolytic anaemia. Br J Haematol. 2013;163(3):393-399. doi:10.1111/bjh.12541
5. Zupańska B, Sylwestrowicz T, Pawelski S. The results of prolonged treatment of autoimmune haemolytic anaemia. Haematologia (Budap). 1981;14(4):425-433.
6. Berentsen S, Barcellini W. Autoimmune Hemolytic Anemias. N Engl J Med. 2021;385(15):1407-1419. doi:10.1056/NEJMra2033982
7. Maskal S, Al Marzooqi R, Fafaj A, et al. Clinical and surgical outcomes of splenectomy for autoimmune hemolytic anemia. Surg Endosc. 2022;36(8):5863-5872. doi:10.1007/s00464-022-09116-x
8. Kuter DJ, Piatek C, Röth A, et al. Fostamatinib for warm antibody autoimmune hemolytic anemia: Phase 3, randomized, double-blind, placebo-controlled, global study (FORWARD). Am J Hematol. 2024;99(1):79-87. doi:10.1002/ajh.27144
9. Fattizzo B, Bortolotti M, Giannotta JA, Zaninoni A, Consonni D, Barcellini W. Intravascular hemolysis and multitreatment predict thrombosis in patients with autoimmune hemolytic anemia. J Thromb Haemost. 2022;20(8):1852-1858. doi:10.1111/jth.15757
10. Roth A, Barcellini W, D'Sa S, et al. Sutimlimab in cold agglutinin disease. N Engl J Med. 2021;384(14):1323-1334. doi: 10.1056/NEJMoa2027760
Key Points
Autoimmune hemolytic anemia is divided into warm antibody hemolytic anemia and cold agglutinin disease based on the temperature at which the autoantibodies react with red blood cells (RBCs).
Hemolysis tends to be more severe in warm antibody hemolytic anemia and can be fatal if reticulocytopenia is also present.
Immunoglobulin and/or complement bound to the patient's RBCs is demonstrated by the occurrence of agglutination after antiglobulin serum is added to washed RBCs (positive direct antiglobulin test).
The pattern of the direct antiglobulin reaction can help distinguish warm antibody hemolytic anemia from cold agglutinin disease.
Treatment is directed at the cause (including stopping medications, avoiding cold, treating the underlying disorder); additional pharmacologic agents depend on the type of hemolytic anemia.
Glucocorticoids remain the first-line treatment for idiopathic warm antibody hemolytic disease, often with rituximab.Glucocorticoids remain the first-line treatment for idiopathic warm antibody hemolytic disease, often with rituximab.





