




Numerose connettiviti causano infiammazione del tratto uveale. (Vedi anche Panoramica sull'uveite.)
Spondiloartropatie
Le spondiloartropatie sieronegative sono una causa frequente di uveite anteriore.
Tra le spondiloartropatie sieronegative, l'infiammazione oculare è più frequente nella spondilite anchilosante, ma si verifica anche in artrite reattiva, malattie infiammatorie intestinali (colite ulcerosa e morbo di Crohn) e artrite psoriasica. L'uveite è classicamente monolaterale, ma sono frequenti le recidive, e l'infiammazione acuta può alternarsi fra i due occhi. Gli uomini sono colpiti più frequentemente delle donne. La maggior parte dei pazienti, a prescindere dal sesso, è positiva per HLA-B27.
La terapia richiede un corticosteroide topico e un farmaco cicloplegico-midriatico. Occasionalmente, sono necessari corticosteroidi iniettati in sede perioculare. Una grave patologia cronica può richiedere l'uso di un farmaco immunosoppressore non-steroideo (p. es., metotrexato o micofenilato mofetile).
Artrite idiopatica giovanile
L'artrite idiopatica giovanile, precedentemente nota come artrite reumatica giovanile, caratteristicamente provoca iridociclite bilaterale cronica nei bambini, in particolare in quelli con la varietà oligoarticolare. Al contrario della maggior parte delle forme di uveite anteriore, tuttavia, l'uveite associata ad artrite idiopatica giovanile tende a non causare dolore, fotofobia e iniezione congiuntivale, ma soltanto offuscamento della vista e miosi, ed è pertanto spesso definita irite bianca. Può essere asintomatica. L'uveite associata ad artrite idiopatica giovanile è più comune nelle giovani donne. Poiché i sintomi possono essere trascurati o assenti, i pazienti con artrite idiopatica giovanile devono essere regolarmente sottoposti a screening.
L'artrite reumatoide, al contrario, non è associata ad uveite isolata ma può causare sclerite, che può causare infiammazione secondaria del tratto uveale.
Crisi infiammatorie ricorrenti sono trattate meglio con un corticosteroide topico e un farmaco cicloplegico-midriatico. Considerati la natura cronica della malattia e il rischio dello sviluppo di una cataratta legata al trattamento o di un glaucoma, il controllo a lungo termine spesso richiede l'uso di un farmaco immunosoppressore non-steroideo (p. es., metotrexato o micofenilato mofetile).
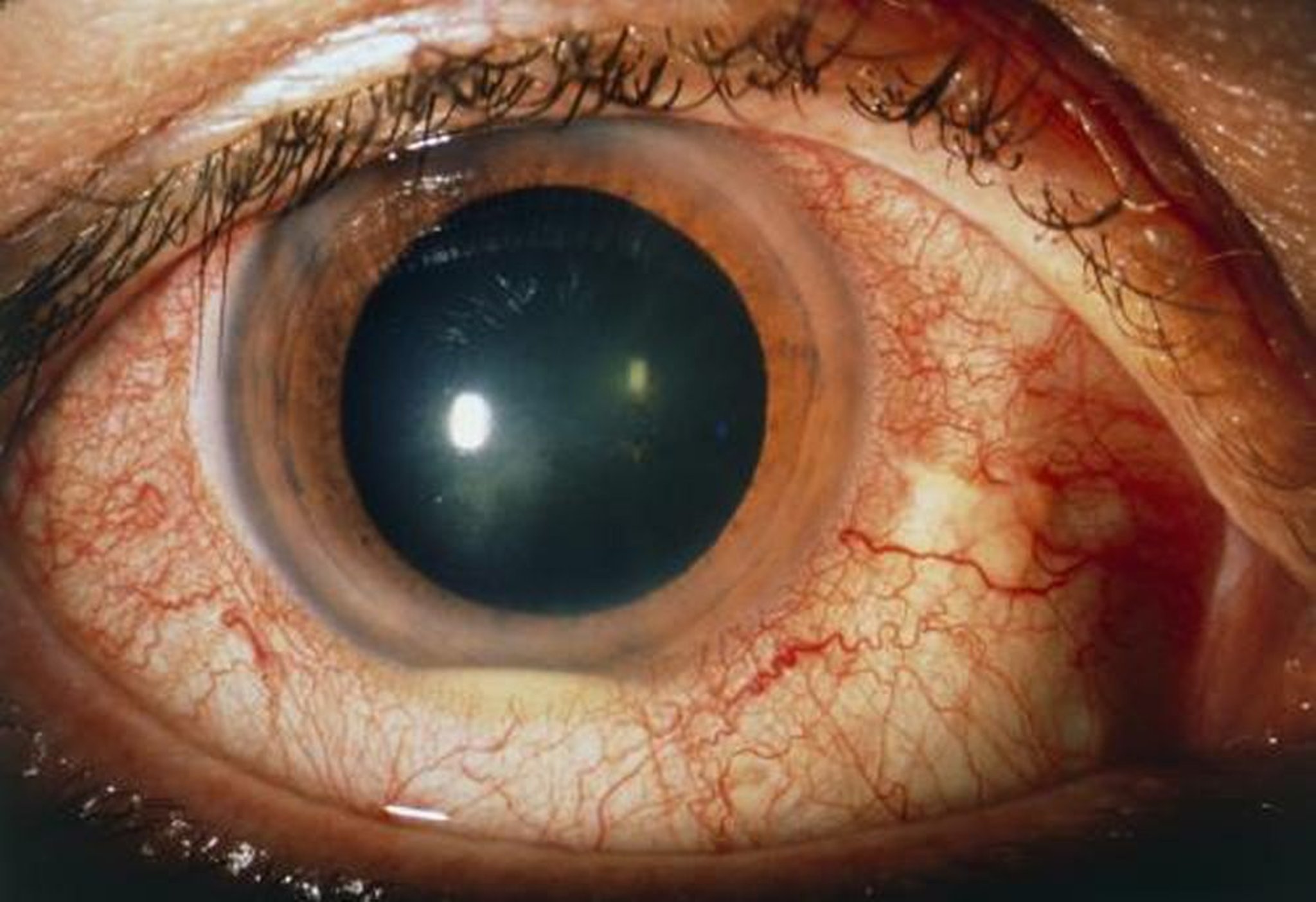
Sarcoidosi
La sarcoidosi è responsabile del 10-20% dei casi di uveite, e circa il 25% dei pazienti con sarcoidosi sviluppa uveite. L'uveite da sarcoidosi è più frequente fra soggetti di discendenza africana e anziani.
Di fatto può verificarsi un qualunque sintomo di uveite anteriore, intermedia, posteriore o panuveite. I reperti indicativi comprendono granulomi congiuntivali, ampi precipitati cheratici sull'endotelio corneale (i cosiddetti precipitati granulomatosi o a grasso di montone), granulomi dell'iride e vasculite retinica. L'indagine bioptica delle lesioni che suggeriscono, che fornisce la diagnosi più sicura, solitamente è effettuata sulla congiuntiva; si effettua raramente su tessuti intraoculari a causa del rischio associato alla procedura.
Il trattamento di solito comporta corticosteroidi topici, perioculari, intraoculari o sistemici, o una loro combinazione, insieme a un farmaco topico cicloplegico-midriatico. I pazienti con infiammazione o malattia da moderata a grave possono richiedere un farmaco immunosoppressore non-steroideo (p. es., metotrexato, micofenolato mofetile).

© Springer Science+Business Media
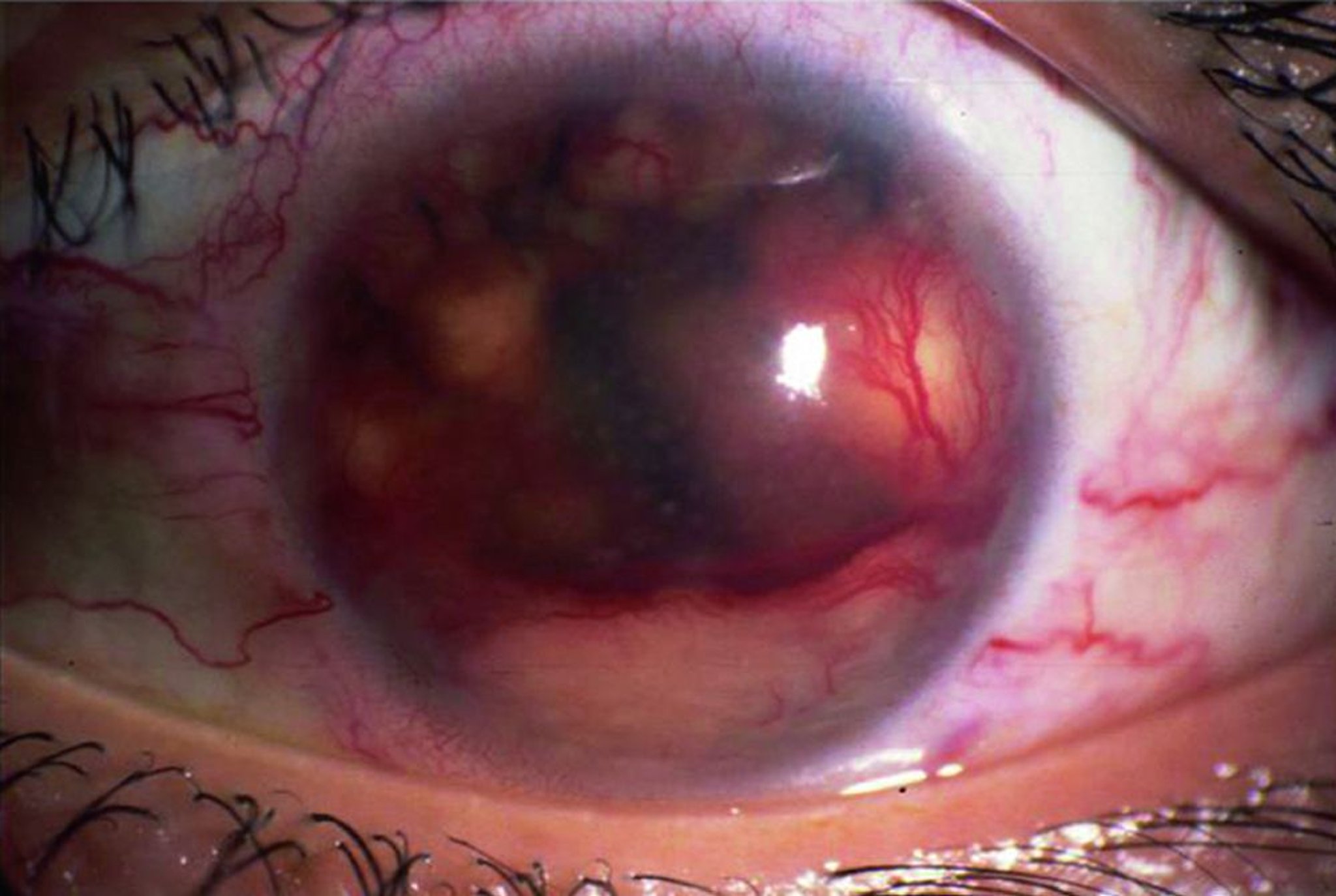
Malattia di Behçet
La malattia di Behçet è rara nel Nordamerica ma è una causa piuttosto frequente di uveite nel Medio Oriente e nell'Estremo Oriente.
I tipici riscontri comprendono grave uveite anteriore con ipopion, retinite, vasculite retinica e infiammazione del disco ottico. Il decorso clinico è generalmente grave, con recidive multiple.
La diagnosi richiede la presenza di manifestazioni sistemiche associate, come ulcere aftose orali o genitali; dermatite, compreso l'eritema nodoso; tromboflebite; o epididimite. Le afte orali possono essere sottoposte a biopsia per ricercare una vasculite occlusiva. Non esistono esami di laboratorio per la malattia di Behçet, ma è associata a HLA-B51.
Il trattamento con corticosteroidi locali e sistemici e un farmaco cicloplegico-midriatico può alleviare le esacerbazioni acute, ma la maggior parte dei pazienti, alla fine, richiede corticosteroidi sistemici e un farmaco immunosoppressore non-steroideo, per controllare l'infiammazione ed evitare le complicanze gravi del trattamento corticosteroideo a lungo termine. Gli agenti biologici, come interferone e inibitori del TNF, si sono mostrati efficaci in pazienti selezionati che non rispondono ad altre terapie. Gli agenti alchilanti (p. es., ciclosporina e clorambucile) hanno indotto la remissione nei pazienti.

SUE FORD/SCIENCE PHOTO LIBRARY
Malattia di Vogt-Koyanagi-Harada
La malattia di Vogt-Koyanagi-Harada è una rara patologia sistemica caratterizzata da uveite accompagnata da anomalie cutanee e neurologiche. La malattia di Vogt-Koyanagi-Harada è particolarmente diffusa tra i soggetti di origine asiatica, indiana, nei discendenti degli indiani d'America e negli Ispanici. Le donne tra i 20 e i 30 anni sono colpite di più degli uomini. L'eziologia è sconosciuta, benché si sospetti fortemente una reazione autoimmune diretta contro cellule contenenti melanina nel tratto uveale, nella cute, nell'orecchio interno e nelle meningi.
I sintomi neurologici tendono a manifestarsi precocemente e comprendono acufeni, disacusia (agnosia uditiva), vertigini, cefalea e meningismo. I riscontri cutanei compaiono spesso più tardi e comprendono vitiligine a chiazze (particolarmente frequente su palpebre, regione lombare e natiche), poliosi (una chiazza localizzata di capelli bianchi, che può coinvolgere le ciglia) e alopecia, che spesso coinvolge il capo e il collo. Riscontri frequenti comprendono distacco sieroso della retina, edema del disco ottico e coroidite. Complicanze a lungo termine comprendono cataratta, glaucoma, fibrosi sottoretinica, e neovascolarizzazione coroideale.
Il trattamento precoce comprende corticosteroidi locali e sistemici e un farmaco cicloplegico-midriatico. Molti pazienti richiedono trattamento con dei farmaci immunosoppressori non-steroidei (p. es., metotrexato, micofenolato mofetile).
Sindrome nefrite tubulointerstiziale e uveite (sindrome TINU [Tubulo Interstitial Nephritis and Uveitis])
La sindrome nefrite tubulointerstiziale e uveite (TINU) tipicamente si presenta con un'uveite anteriore bilaterale acuta non granulomatosa, sebbene sia frequentemente accompagnata da complicanze posteriori, tra cui l'edema del nervo ottico e della macula. Colpisce più comunemente le femmine adolescenti, ma può presentarsi a qualsiasi età. I sintomi comprendono dolore oculare e arrossamento, riduzione dell'acuità visiva e fotofobia. I pazienti possono avere un'anamnesi di sintomi da prodromi virali e anche dolore al fianco, poliuria e nicturia. La valutazione per la nefrite può includere i livelli della beta-2 microglobulina e a volte una biopsia renale. Mentre il danno renale acuto è frequentemente autolimitato nella sindrome nefrite tubulointerstiziale e uveite (TINU), la malattia oculare è spesso cronica (1).
Il trattamento con steroidi orali nella fase acuta e l'uso di farmaci immunosoppressori non steroidei (p. es., micofenolato mofetile) per la malattia cronica è spesso necessario.
Riferimento per la sindrome nefrite tubulointerstiziale e uveite (TINU)
1. Koreishi AF, Zhou M, Goldstein DA: Tubulointerstitial nephritis and uveitis syndrome: Characterization of clinical features. Ocul Immunol Inflamm 17;29(7-8):1312-1317, 2021. doi: 10.1080/09273948.2020.1736311







