




Die dilatative Kardiomyopathie ist eine myokardiale Dysfunktion, die eine Herzinsuffizienz mit führender ventrikulärer Dilatation und systolischer Dysfunktion hervorruft. Zu den Symptomen gehören Dyspnoe, Müdigkeit und periphere Ödeme. Die Diagnose erfolgt klinisch und durch erhöhte natriuretische Peptide, Thoraxröntgen, Echokardiographie und MRT. Die Behandlung richtet sich nach der jeweiligen Ursache. Wenn die Herzinsuffizienz progressiv und schwer ist, kann eine kardiale Resynchronisationstherapie, ein implantierbarer Kardioverter-Defibrillator, eine Reparatur moderater bis schwerer Klappeninsuffizienz, ein linksventrikuläres Herzunterstützungssystem oder eine Herztransplantation notwendig sein.
Eine Kardiomyopathie ist eine primäre Störung des Herzmuskels (siehe auch Übersicht der Kardiomyopathien). Die dilatative Kardiomyopathie kann sich in jedem Alter entwickeln, tritt aber häufiger bei Erwachsenen auf, die jünger als etwa 50 Jahre alt sind. Etwa 10% der Menschen, die eine dilatative Kardiomyopathie entwickeln, sind älter als 65 Jahre. In den Vereinigten Staaten tritt die Krankheit bei etwa 3-mal so vielen Männern wie Frauen und 3-mal so vielen Menschen afrikanischer Abstammung wie Weißen auf. Etwa 5 bis 8 von 100.000 Menschen entwickeln die Erkrankung jedes Jahr (1).
Allgemeiner Hinweis
1. Dec GW, Fuster V: Idiopathic dilated cardiomyopathy. N Engl J Med 331:1564–1575, 1994.
Pathophysiologie der dilatativen Kardiomyopathie
Als primäre Herzmuskelerkrankung tritt die myokardiale Dysfunktion der dilatativen Kardiomyopathie auf, wenn keine anderen Erkrankungen vorliegen, die ein dilatatives Myokard verursachen können, wie z. B. eine schwere koronare Verschlusskrankheit oder Zustände, die eine Druck- oder Volumenüberladung des Ventrikels mit sich bringen (z. B. Hypertonie, Herzklappenerkrankung). Bei manchen Patienten wird angenommen, dass die dilatative Kardiomyopathie mit einer akuten Myokarditis beginnt (wahrscheinlich meist viraler Genese), gefolgt von einer unterschiedlich langen latenten Phase, einer Phase mit diffuser Nekrose der Kardiomyozyten (aufgrund einer Autoimmunreaktion auf viral veränderte Myozyten) und einer chronischen Fibrose. Unabhängig von der Ursache dilatiert das Myokard, dünnt aus und hypertrophiert kompensatorisch (siehe Abbildung Formen der Kardiomyopathie), was häufig zu einer funktionellen Mitralklappeninsuffizienz und/oder Trikuspidalklappeninsuffizienz und Vorhofvergrößerung führt.
Die Krankheit betrifft bei den meisten Patienten beide Ventrikel, bei wenigen auch nur den LV und selten nur den RV.
Wenn die Kammerdilatation und -dysfunktion signifikant ist, können sich aufgrund der Blutstauung murale Thromben bilden. Kardiale Arrhythmien und atrioventrikuläre Blockierungen komplizieren oft die akute Myokarditis und die späte Phase der Dilatation. Vorhofflimmern tritt häufig auf, wenn der linke Vorhof dilatiert ist.
Ätiologie der dilatativen Kardiomyopathie
Die dilatative Kardiomyopathie hat viele bekannte und wahrscheinlich auch viele unbekannte Ursachen (siehe Tabelle Ursachen der dilatativen Kardiomyopathie). Mehr als 20 Viren können eine dilatative Kardiomyopathie hervorrufen; in gemäßigten Klimazonen ist das Coxsackie-B-Virus die häufigste virale Ursache. In Mittel- und Südamerika ist die Chagas-Krankheit aufgrund von Trypanosoma cruzi die häufigste Infektionsursache.
Andere Ursachen sind verlängerte (chronische) Tachykardie, HIV-Infektion, Toxoplasmose, Thyrotoxikose und Beriberi. Viele toxische Substanzen, v. a. Alkohol, verschiedene organische Lösungsmittel, Eisen- oder Schwermetallionenund bestimmte Chemotherapeutika (z. B. Doxorubicin, Trastuzumab), schädigen das Herz. Häufige ventrikuläre Ektopien (> 10.000 ventrikuläre Extrasystolen/Tag) wurden mit einer linksventrikulären systolischen Dysfunktion in Verbindung gebracht.
Plötzlicher emotionaler Stress und andere hyperadrenerge Zustände können akute dilatative Kardiomyopathie auslösen, die typischerweise reversibel ist (ebenso die durch längere Tachykardie verursachte DCM). Ein Beispiel ist die akute apikale Ballon-Kardiomyopathie (auch Takotsubo-Kardiomyopathie, Stress-Kardiomyopathie oder gebrochenes Herz-Syndrom genannt). Bei dieser Erkrankung sind in der Regel der Apex und gelegentlich auch andere Segmente des linken Ventrikels betroffen, was zu einer regionalen Wandstörung und manchmal zu einer fokalen Dilatation (Ballonbildung) führt.
Genetische Faktoren spielen in 20–35% der Fälle eine Rolle; bisher wurden > 60 Gene und Genabschnitte impliziert.
Symptome und Zeichen der dilatativen Kardiomyopathie
Der Beginn der dilatativen Kardiomyopathie ist in der Regel allmählich, außer bei akuter Myokarditis, akuter apikaler Ballonkardiomyopathie und Tachyarrhythmie-induzierter Kardiomyopathie. Etwa 25% aller Patienten mit dilatativer Kardiomyopathie haben atypische Brustschmerzen. Andere Symptome hängen davon ab, welcher Ventrikel betroffen ist.
Eine linksventrikuläre Dysfunktion verursacht Dyspnoe und Erschöpfung bei Anstrengung aufgrund eines erhöhten linksventrikulären diastolischen Drucks und eines erniedrigen Herzzeitvolumens.
Rechtsventrikuläre Insuffizienz verursacht periphere Ödeme und einen Halsvenenstau. Selten ist der RV besonders bei jüngeren Patienten betroffen; dann sind Vorhofrhythmusstörungen und ein plötzlicher Herztod aufgrund maligner ventrikulärer Tachyarrhythmien typisch.
Diagnose der dilatativen Kardiomyopathie
Röntgenthorax
EKG
Echokardiographie
Herz-MRT
Endomyokardbiopsie (ausgewählte Fälle)
Tests auf Ursachen falls indiziert
Die Diagnose der dilatativen Kardiomyopathie erfolgt durch Anamnese, körperliche Untersuchung und Ausschluss anderer häufiger Ursachen des Herzversagens (z. B. systemischer Bluthochdruck, primäre Herzklappenerkrankungen, Myokardinfarkt, siehe Tabelle Diagnose und Behandlung von Kardiomyopathien). Insbesondere bei dilatativer Kardiomyopathie ohne eindeutige Ursache sollte eine sorgfältige Familienanamnese erhoben werden, um Familienmitglieder mit möglicherweise früh einsetzender Herzerkrankung, Herzinsuffizienz, oder plötzlichem Tod zu identifizieren. In vielen Zentren werden Familienmitglieder ersten Grades auf kardiale Funktionsstörungen untersucht (z. B. mittels Echokardiographie). Da andere häufige Ursachen einer ventrikulären Insuffizienz ausgeschlossen werden müssen, sind Röntgenthorax, EKG, Echokardiographie und Herz-MRT erforderlich. Eine Endomyokardbiopsie wird in vereinzelten Fällen durchgeführt
Serum-Herzmarker werden gemessen, wenn akute Symptome oder Brustschmerzen vorhanden sind. Obwohl dies typisch für eine koronare Ischämie ist, tritt die Troponinerhöhung häufig bei Herzinsuffizienz auf, insbesondere wenn die Nierenfunktion vermindert ist. Die Level des natriuretischen Peptids i. S. sind typischerweise erhöht, wenn Herzinsuffizienz vorliegt.
Klinisch vermutete spezifische Ursachen werden diagnostiziert (siehe an anderer Stelle im MSD-Manual). Wenn aus klinischer Sicht keine spezifische Ursache vorliegt, werden in entsprechenden Fällen das Serumferritin, die Eisenbindungskapazität und die TSH-Spiegel bestimmt.
Serologische Tests für Toxoplasma, T. cruzi, Coxsackievirus und Echovirus können in angemessenen Fällen durchgeführt werden.
Das Röntgenthoraxbild zeigt üblicherweise eine Kardiomegalie aller Herzkammern. Ein Pleuraerguss, speziell der rechten Seite, begleitet häufig die erhöhten pulmonalvenösen Drücke und ein interstitielles Ödem.

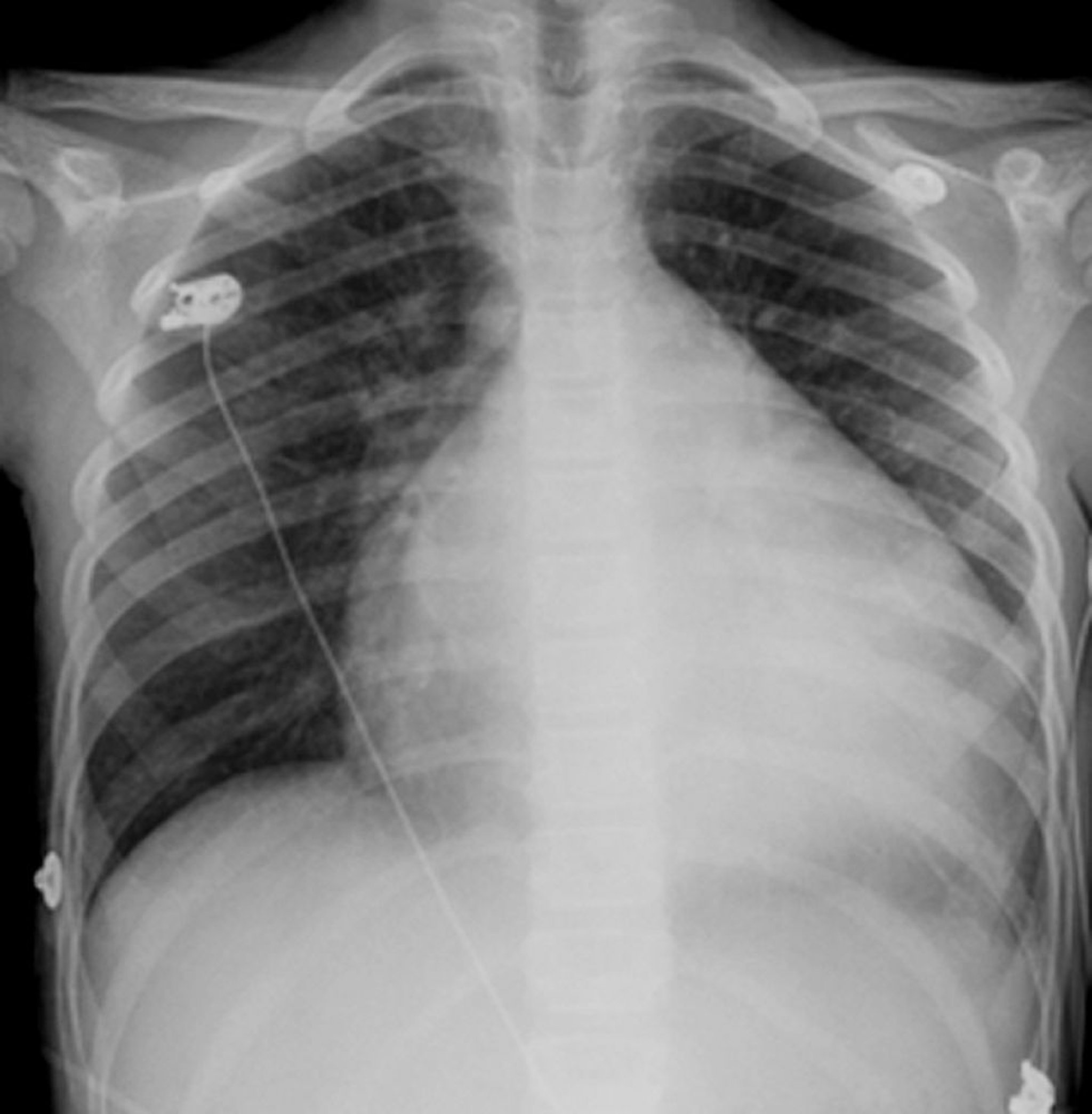
© Springer Science+Business Media
Das EKG kann eine Sinustachykardie und unspezifische ST-Strecken-Senkungen mit Niedervoltage und invertierten T-Wellen zeigen. Manchmal sind pathologische Q-Zacken in den präkordialen Ableitungen vorhanden, die einen vorangegangenen Myokardinfarkt simulieren. Ein Linksschenkelblock und Vorhofflimmern sind häufig.
Die Echokardiographie zeigt die dilatierten, hypokinetischen Herzkammern und schließt primäre Herzklappenkrankheiten aus. Segmentale Wandbewegungsstörungen können auch bei der dilatativen Kardiomyopathie auftreten, da der Prozess lückenhaft sein kann. Die Echokardiographie kann auch einen wandständigen Thrombus zeigen.
Ein kardiales MRT wird immer häufiger angefertigt und ist nützlich, um eine genaue Bildgebung der Herzstruktur oder -funktion zu liefern. Die MRT mit Gadoliniumkontrastmittel kann eine abnorme Herzmuskelgewebetextur oder Narbenmuster zeigen (z. B. späte Gadoliniumsteigerung oder LGE). Das Muster von LGE kann bei aktiver Myokarditis, Sarkoidose, Muskeldystrophie oder Chagas-Krankheit diagnostisch sein.

© 2017 Elliot K. Fishman, MD.
Positronen-Emissions-Tomographie hat sich als sensitiv bei der Diagnose von Herzsarkoidose erwiesen.
Eine Koronarangiographie kann erforderlich sein, um eine Koronararterienerkrankung als Ursache einer LV-Dysfunktion auszuschließen, wenn die Diagnose nach nichtinvasiven Tests in Frage gestellt wird. Patienten mit Brustschmerzen oder mehreren kardiovaskulären Risikofaktoren sowie ältere Patienten haben häufiger eine koronare Herzkrankheit. Beide Ventrikel können während der Katheterisierung in ausgewählten Fällen biopsiert werden, in denen die Ergebnisse das Management verändern.
Eine Endomyokardbiopsie ist indiziert, wenn eine Riesenzellmyokarditis, eosinophile Myokarditis oder Sarkoidose vermutet wird, da die Ergebnisse die Therapie beeinflussen.
Prognose für dilatative Kardiomyopathie
Die Prognose der dilatativen Kardiomyopathie ist im Allgemeinen schlecht, obwohl sie sich mit den heutigen Behandlungsmethoden (z. B. Einsatz von Betablockern, Angiotensin-Converting-Enzyme [ACE]-Hemmern, Mineralocorticoid-Rezeptor-Antagonisten, Natrium-Glukose-Cotransporter-2-Protein (SGLT2)-Hemmern, implantierbaren Kardioverter-Defibrillatoren oder kardialer Resynchronisationstherapie) verbessert hat. Ungefähr 20% Patienten sterben im ersten Jahr, danach etwa 10%/Jahr; etwa 40–50% der Todesfälle treten plötzlich, aufgrund einer malignen Herzrhythmusstörung oder einer Embolie, auf. Die Prognose ist für Frauen besser als für Männer, und Menschen afrikanischer Abstammung überleben etwa halb so lange wie Weiße.
Die Prognose ist besser, wenn eine kompensatorische Hypertrophie die ventrikuläre Wanddicke aufrechterhält, und ist schlechter, wenn die Ventrikelwand erheblich ausdünnt und der Ventrikel dilatiert. Patienten, bei denen die dilatative Kardiomyopathie durch die Behandlung gut kompensiert wird, können viele Jahre stabil sein.
Behandlung der dilatativen Kardiomyopathie
Ursache (falls vorhanden) behandelt
Standardtherapie bei Herzversagen mit reduziertem Auswurffraktion
Antikoagulanzien, wenn Vorhofflimmern oder eine andere Indikation vorliegt
Manchmal implantierbarer Kardioverter-Defibrillator, kardiale Resynchronisationstherapie (CRT), linksventrikuläres Herzunterstützungssystem oder Transplantation
Immunsuppression bei Patienten mit Riesenzellmyokarditis, eosinophiler Myokarditis oder Sarkoidose
Behandelbare Ursachen (z. B. Toxoplasmose, akute Chagas-Krankheit, Hämochromatose, Thyreotoxikose, Beriberi) werden behoben. Patienten mit HIV-Infektion sollten antiretrovirale Therapie (ART) optimiert. Die Behandlung mit Immunsuppression sollte auf Patienten mit Biopsie-nachgewiesener Riesenzellmyokarditis, eosinophiler Myokarditis oder Sarkoidose beschränkt sein.
Ansonsten ist die Behandlung die gleiche wie bei Herzinsuffizienz mit verminderter Ejektionsfraktion: ACE-Hemmer, Betablocker, Aldosteronrezeptorblocker, Angiotensin-II-Rezeptorblocker, ARNI (Angiotensin-II-Rezeptorblocker und Neprilysin-Inhibitor), Natrium-Glukose-Cotransporter-2-Protein-Hemmer (SGLT), Hydralazin/Nitrate, Diuretika und Digoxin. Studien haben gezeigt, dass Patienten mit idiopathischer dilatativer Kardiomyopathie besonders gut auf die Standardbehandlung der Herzinsuffizienz ansprechen und im Allgemeinen besser abschneiden als Patienten mit ischämischer Herzkrankheit.
Besondere Vorsichtsmaßnahmen sind bei der Behandlung der peripartalen Kardiomyopathie erforderlich. Viele Medikamente (z. B. ACE-Hemmer und Angiotensin-II-Rezeptor-Blocker) sollten während der Schwangerschaft wegen des Risikos einer fetalen Schädigung vermieden werden. Darüber hinaus werden diese Medikamente für stillende Frauen nicht empfohlen.
Prophylaktische orale Antikoagulation wurde in der Vergangenheit zur Vorbeugung von Wandthromben bei anderen Formen der Kardiomyopathie eingesetzt. Die Verwendung von Antikoagulanzien bei Patienten mit eingeschränkter linksventrikulärer Funktion und im Sinusrhythmus ist nach wie vor umstritten, und die Verwendung von Antikoagulanzien in dieser Situation ist keine Routine. Warfarin oder ein direkes orales Antikoagulans (DOAC) wird empfohlen, wenn eine spezifische Indikation vorliegt (z. B. frühere zerebrovaskuläre Embolie, identifizierter Herz-Thrombus, Vorhofflimmern und/oder Flattern).
Sowohl die Leitlinien der American Heart Association als auch der European Society of Cardiology empfehlen, eine Antikoagulationstherapie bei Patienten mit peripartaler Kardiomyopathie in Betracht zu ziehen, die aufgrund des Risikos eines hyperkoagulierbaren Zustands während der Schwangerschaft sehr niedrige Ejektionsfraktionen aufweisen (1, 2). Es wurde niedermolekulares Heparin verwendet. Warfarin und wahrscheinlich auch direkt wirkende orale Antikoagulanzien sollten jedoch in bestimmten Phasen der Schwangerschaft wegen des fetalen Risikos nicht verwendet werden.
Die medizinische Behandlung von Herzinsuffizienz reduziert das Risiko von Herzrhythmusstörungen, aber einimplantierbarer Kardioverter-Defibrillator kann bei Patienten, die trotz optimaler medikamentöser Therapie weiterhin eine reduzierte Ejektionsfraktion aufweisen, eingesetzt werden, um Todesfällen aufgrund von plötzlichen Herzrhythmusstörungen vorzubeugen. Da sich die atrioventrikuläre (AV) Blockade bei akuter Myokarditis oft auflöst, ist ein permanenter Herzschrittmacher in der Regel nicht notwendig. Ein permanenter Herzschrittmacher kann jedoch erforderlich sein, wenn der AV-Block während der chronisch erweiterten Phase andauert oder sich entwickelt. Wenn Patienten trotz optimaler medizinischer Behandlung ein erweitertes QRS-Intervall mit niedriger linksventrikulärer Ejektionsfranktion und schweren Symptomen aufweisen, sollte eine kardiale Resynchronisationstherapie in Betracht gezogen werden.
Patienten mit refraktärem Herzversagen trotz Therapie können Kandidaten für eine Herztransplantation werden. Die Auswahlkriterien umfassen das Fehlen einer assoziierten systemischen Krankheit, psychiatrische Krankheiten und einen irreversibel erhöhten pulmonarteriellen Gefäßwiderstand; da Spenderherzen rar sind, wird jüngeren Patienten (üblicherweise < 70 Jahre) die höhere Priorität eingeräumt. Linksventrikuläre Herzunterstützungssysteme können bei einigen Patienten (z. B. Patienten, die nicht für eine Herztransplantation in Frage kommen) auch als eine Brücke zur Herztransplantation oder als Zieltherapie in Betracht gezogen werden. In der Zieltherapie wird ein LVAD als permanente Therapie für Patienten mit refraktärer Herzinsuffizienz eingesetzt (und nicht als vorübergehende Maßnahme vor einer Herztransplantation).
Literatur zur Behandlung
1. Bozkurt B, Colvin M, Cook J, et al: Current diagnostic and treatment strategies for specific dilated cardiomyopathies: A Scientific Statement From the American Heart Association. Circulation 134(23):e579–e646.2, 2016.
2. Regitz-Zagrosek V, Roos-Hesselink JW, Bauersachs J, et al: 2018 ESC Guidelines for the management of cardiovascular diseases during pregnancy: The Task Force for the Management of Cardiovascular Diseases during Pregnancy of the European Society of Cardiology (ESC). Eur Heart J 39: 3165–3241, 2018.
Wichtige Punkte
Bei dilatativer Kardiomyopathie kommt es zu einer Dilatation, Verdünnung und Hypertrophie des Myokards.
Ursachen sind Infektionen (häufig viral), Toxine und Stoffwechsel-, genetische oder Bindegewebserkrankungen.
Führen Sie Thoraxröntgen, EKG, Echokardiographie und Herz-MRT durch, um das Ausmaß der Erkrankung und die endomyokardiale Biopsie bei ausgewählten Patienten zu beurteilen.
Suchen Sie nach anderen Ursachen für die Herzinsuffizienz.
Behandeln Sie, wenn möglich, die primäre Ursache und verwenden Sie Standardmaßnahmen zur Behandlung der Herzinsuffizienz (z. B. Angiotensin-Converting-Enzym [ACE]-Hemmer, Betablocker, Aldosteron-Rezeptorblocker, Angiotensin-II-Rezeptorblocker, ARNI [Angiotensin-II-Rezeptorblocker und Neprilysin-Inhibitor], Natrium-Glukose-Cotransporter-2-Protein (SGLT2)-Hemmer, Hydralazin/Nitrate, Diuretika, Digoxin, implantierbarer Kardioverter-Defibrillator und/oder kardiale Resynchronisationstherapie).
Verwenden Sie bei ausgewählten Patienten orale Antikoagulanzien und Immunsuppressiva.
Weitere Informationen
Die folgenden englischsprachigen Quellen können nützlich sein. Bitte beachten Sie, dass das MSD-Manual nicht für den Inhalt dieser Quellen verantwortlich ist.
American Heart Association guidelines on dilated cardiomyopathies: Bozkurt B, Colvin M, Cook J, et al: Current diagnostic and treatment strategies for specific dilated cardiomyopathies: A Scientific Statement From the American Heart Association. Circulation 134(23):e579–e646.2, 2016.
European Society of Cardiology guidelines on cardiomyopathy in pregnant patients: Regitz-Zagrosek V, Roos-Hesselink JW, Bauersachs J, et al: 2018 ESC Guidelines for the management of cardiovascular diseases during pregnancy: The Task Force for the Management of Cardiovascular Diseases during Pregnancy of the European Society of Cardiology (ESC). European Heart Journal 39: 3165–3241, 2018.