




Die Autoimmunmyositis ist durch entzündliche und degenerative Veränderungen in den Muskeln (Polymyositis, nekrotisierende immunvermittelte Myopathie) oder in Haut und Muskeln (Dermatomyositis) gekennzeichnet. Manifestationen sind symmetrische Schwäche, gelegentlich Empfindlichkeit und faseriger Muskelersatz, manchmal mit Atrophie, hauptsächlich der proximalen Gürtelmuskulatur. Die Diagnose wird anhand von klinischen Befunden und Anomalien bei Muskeltests gestellt, zu denen Kreatinkinase-Test, MRT, Elektromyographie und Muskelbiopsie gehören können. Mehrere Arten von Myositis haben pulmonale und kardiale Manifestationen. Die Therapie besteht aus Kortikosteroiden, in Kombination mit Immunsuppressiva und/oder IV Immunglobulin.
Die Autoimmunmyositis tritt bei Frauen häufiger auf als bei Männern, und zwar im Verhältnis 2:1. Die Inzidenz ist bei Schwarzen 3- bis 4-mal höher als bei Weißen. Ein Auftreten ist in jedem Alter möglich, am häufigsten zwischen dem 40. und dem 60. Lebensjahr, im Kindesalter zwischen dem 5. und 15.
Ätiologie der Autoimmunmyositis
Die Ursache von autoimmuner Myositis scheint eine Autoimmunreaktion des Muskelgewebes bei genetisch anfälligen Menschen zu sein. Es kommt zu einer familiären Häufung, und die Subtypen der menschlichen Leukozytenantigene (HLA) sind mit Myositis assoziiert. Zum Beispiel erhöhen die Allele des Vorfahren-Haplotyps 8.1 (HLA-DRB1*03-DQA1*05-DQB1*02) das Risiko für Polymyositis, Dermatomyositis und interstitielle Lungenerkrankungen. Als Auslöser fungieren möglicherweise virale Myositiden und begleitende Krebserkrankungen. Die bekannte Assoziation der Dermatomyositis (weniger der Polymyositis) mit Krebserkrankungen legt die Vermutung nahe, dass Tumoren eine Myositis als Folge einer Autoimmunreaktion auf ein gemeinsames Antigen in Muskel und Tumor induzieren können.
Pathophysiology of Autoimmune Myositis
Zu den typischen pathologischen Veränderungen zählen zelluläre Schädigung und Atrophie mit unterschiedlichem Entzündungsausmaß. Die Muskulatur von Händen, Füßen und Gesicht ist weit weniger betroffen als die übrige Skelettmuskulatur. Bei Befall von Muskeln im Pharynx, dem oberen Ösophagus sowie gelegentlich dem Herz kann es zur Beeinträchtigung der Organfunktion kommen. Entzündliche Prozesse können auch in Gelenken und Lungen auftreten, speziell bei Patienten mit Antisynthetaseantikörpern.
Dermatomyositis ist gekennzeichnet durch Immunkomplexablagerungen in den Gefäßen und gilt als eine komplementvermittelte Vaskulopathie. Im Gegensatz dazu ist die Polymyositis durch eine direkte T-Zell-vermittelte Muskelschädigung gekennzeichnet, und nekrotisierende immunvermittelte Myopathien sind durch Makrophageninfiltrate und Myophagozytose gekennzeichnet.
Klassifikation der autoimmunen Myositis
Eine autoimmune Myositis kann in 4 Gruppen eingeteilt werden, hauptsächlich basierend auf Histopathologie und klinischer Präsentation:
Polymyositis
Dermatomyositis
Nekrotisierende immunvermittelte Myopathien
Einschlusskörpermyositis
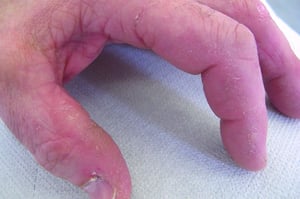
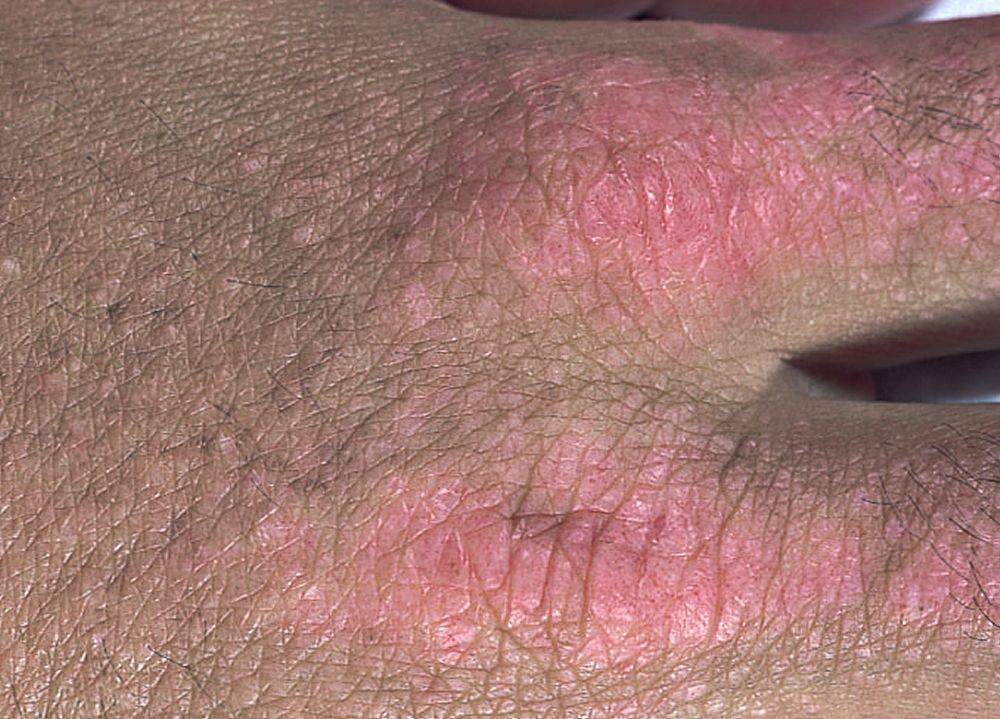
Die Dermatomyositis kann durch den charakteristischen Hautbefunde der Dermatomyositis von Polymyositis unterschieden werden (vgl. Symptome und Zeichen). Die Histopathologie der Muskeln unterscheidet sich ebenfalls. Dermatomyositis und Polymyositis können sich als reine Muskelerkrankungen oder als Teil des Antisynthetasesyndroms manifestieren, wenn sie mit Arthritis (normalerweise nicht erosiv), Fieber, interstitielle Lungenerkrankung, Hyperkeratose der radialen Seite der Finger (Hände des Mechanikers) und Raynaud-Syndrom in Zusammenhang stehen.
Nekrotisierende, immunvermittelte Myopathien umfassen am häufigsten Signalerkennungspartikel- (SRP) -Antikörper-bedingte Myositis und Statin-induzierte Myositis, haben gewöhnlich eine aggressive Präsentation, haben sehr erhöhte Kreatinkinasespiegel (CK) und betreffen keine extramuskulären Organe (1).
Die Einschlusskörpermyositis verursacht eine proximale Beinmuskelschwäche, betrifft aber häufig auch die distalen Muskeln (z. B. Hand- und Fußmuskeln) und führt häufig zu Muskelschwund. Sie entwickelt sich im höheren Lebensalter, verläuft langsamer und spricht im Allgemeinen nicht auf eine immunsuppressive Therapie an.
Die Autoimmunmyositis kann sich auch mit anderen autoimmunen rheumatischen Erkrankungen überschneiden — z. B. systemischer Lupus erythematodes, systemische Sklerose, verschiedene Erkrankungen des Bindegewebe. Diese Patienten weisen neben Myositis (manifest sich entweder als Dermatomyositis oder Polymyositis) Symptome und Zeichen der Überlappungsstörung auf.
Hinweis zur Klassifizierung
1. Lundberg IE, Fujimoto M, Vencovsky J, et al. Idiopathic inflammatory myopathies. Nat Rev Dis Primers 7(1):86, 2021. doi:10.1038/s41572-021-00321-x
Symptome und Anzeichen einer Autoimmunmyositis
Der Beginn der Autoimmunmyositis kann akut (vor allem bei Kindern) oder heimtückisch (vor allem bei Erwachsenen) sein. Polyarthralgien, Raynaud-Syndrom, Dysphagie, pulmonale Symptome (z. B. Husten, Dyspnoe) und Allgemeinbeschwerden (v. a. Fieber, Müdigkeit und Gewichtsverlust) können zusätzlich auftreten. Eine schwere Erkrankung ist gekennzeichnet durch Dysphagie, Dysphonie und/oder Zwerchfellschwäche.
Die Muskelschwäche kann über Wochen bis Monate fortschreiten. Es bedarf jedoch der Destruktion von 50% der Muskelfasern, um eine symptomatische Muskelschwäche hervorzurufen, d. h., diese Manifestation signalisiert eine bereits fortgeschrittene Myositis. Die Patienten schildern Schwierigkeiten beim Heben der Arme über die Schultern, beim Treppensteigen oder beim Aufstehen aus der Sitzposition. Manchmal entwickeln sich Muskelempfindlichkeit und Atrophie. Patienten können den Gebrauch eines Rollstuhls benötigen oder wegen der Schwäche der Becken- und Schultergürtelmuskulatur bettlägerig werden. Die Nackenbeugemuskeln können so schwer betroffen sein, dass der Kopf nicht mehr vom Kissen gehoben werden kann. Der Befall der Pharynx- und oberen Speiseröhrenmuskulatur kann das Schlucken erschweren und zu Aspiration prädisponieren. Muskeln der Hände, Füße und des Gesichts sind nicht beteiligt, außer bei der Einschlusskörper-Myositis, bei der eine distale Beteiligung, insbesondere der Hände, charakteristisch ist. Kontrakturen der Extremitäten treten selten auf.
Gelenkbeteiligung gehören Polyarthralgie oder Polyarthritis mit Schwellungen und anderen Merkmalen einer nichtdestruktiven Arthritis. Sie ist am häufigsten bei einer durch anti-Jo-1- oder andere Antisynthetaseantikörper gekennzeichneten Sonderform zu finden.
Abgesehen von Beteiligung des Pharynx und oberen Ösophagus ist eine Organbeteiligung bei der autoimmune Myositis deutlich seltener zu finden als bei anderen rheumatischen Systemkrankheiten, wie z. B. dem systemischen Lupus erythematodes oder der systemischen Sklerose. Insbesondere bei Patienten mit Antisynthetaseantikörpern kann eine interstitielle Lungenerkrankung im Vordergrund stehen. Eine kardiale Beteiligung, insbesondere Erregungsleitungsstörungen und ventrikuläre Dysfunktion, kann auftreten. Bei Kindern mit Begleitvaskulitis können gastrointestinale Symptome wie Hämatemesis, Meläna und ischämische Darmperforation hinzukommen.
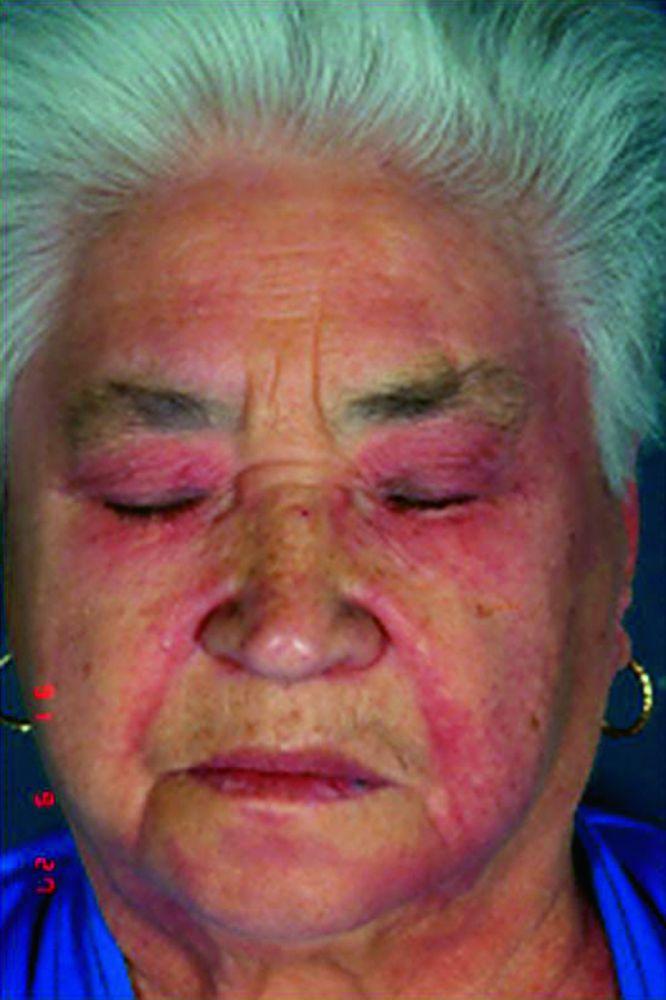
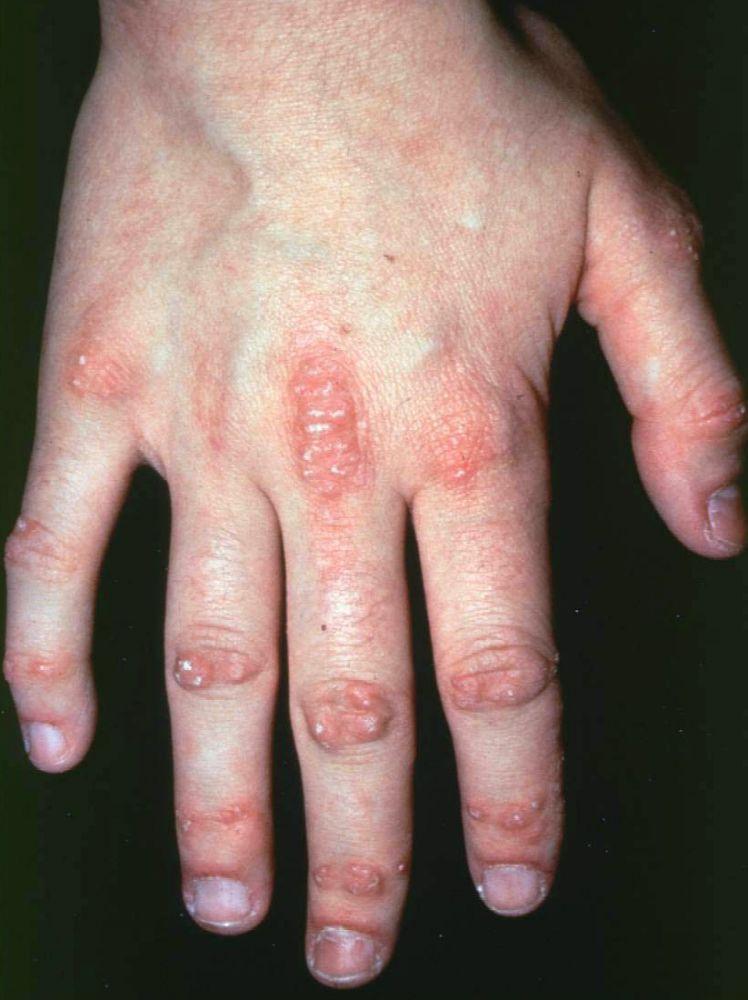
Die bei der Dermatomyositis auftretenden Hautveränderungen haben einen dunklen erythematösen Charakter. Lichtempfindlichkeit und Hautgeschwüre sind sichtbar. Ein purpurfarbenes periorbitales Ödem (heliotropischer Ausschlag) ist für diese Krankheit relativ spezifisch. Anderswo kann der Hautausschlag leicht erhaben und glatt oder schuppig auftreten; er zeigt sich an der Stirn, V-förmig im Nacken-Schulter-Bereich, am Thorax oder Rücken, an Unterarmen und Unterschenkeln, seitliche Oberschenkel, an Ellbogen- und Kniegelenken, den medialen Malleoli sowie an den dorsalen Seiten der proximalen Interphalangealgelenke (PIP) und der Fingergrundgelenke (MCP) (Gottron-Papeln – ein relativ spezifischer Befund). Basis und Seiten der Fingernägel können hyperämisch oder verdickt sein. Über den radialen Seiten der Finger kann eine Dermatitis mit Abschilferung und Rissen auftreten. Subkutane und Muskelkalkeinlagerungen sind v. a. bei betroffenen Kindern zu finden. Die primären Hautläsionen klingen häufig vollständig ab, können aber sekundäre Veränderungen nach sich ziehen (z. B. bräunliche Pigmentierung, Atrophie, persistierende Neovaskularisierung, Narbenbildung). Ein Ausschlag auf der Kopfhaut erscheint psoriasisförmig und stark juckend.
Charakteristische Hautveränderungen können auftreten, wenn keine Muskelerkrankung vorliegt. In diesem Fall wird die Krankheit als amyopathische Dermatomyositis bezeichnet.
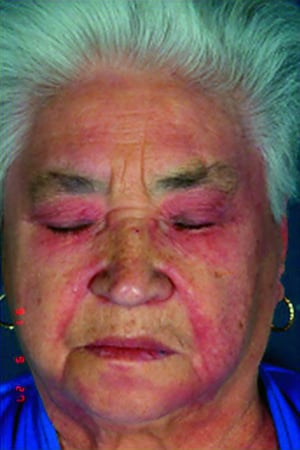
© Springer Science+Business Media
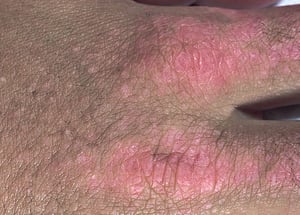
© Springer Science+Business Media
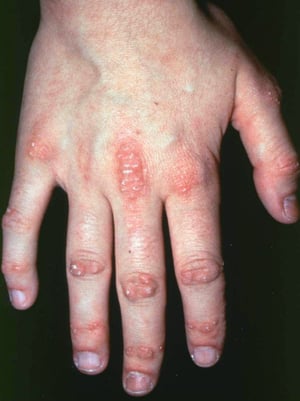
© Springer Science+Business Media
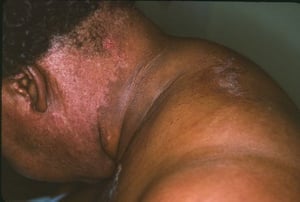
Image courtesy of Karen McKoy, MD.
© Springer Science+Business Media

© Springer Science+Business Media

© Springer Science+Business Media

© Springer Science+Business Media

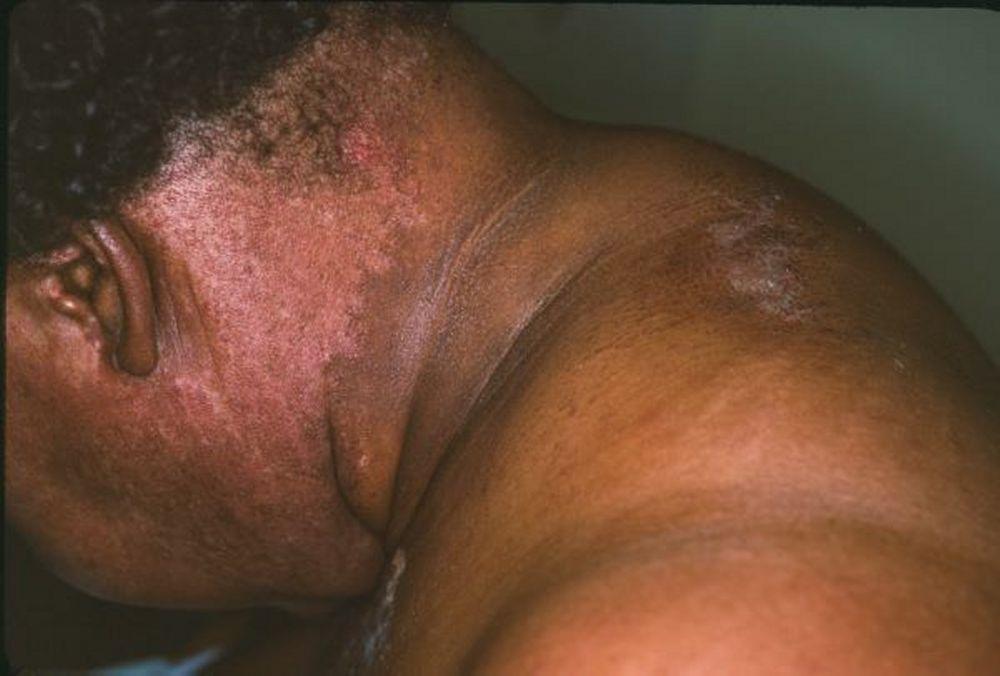
Image courtesy of Karen McKoy, MD.

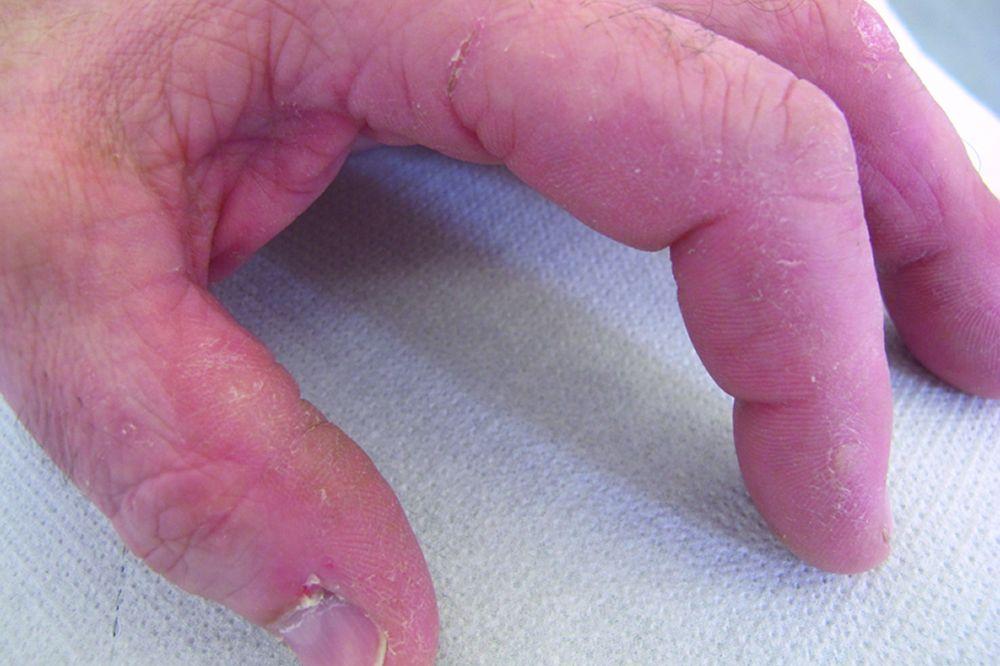
© Springer Science+Business Media
Diagnose von Autoimmunmyositis
Klinische Kriterien
Muskelbiopsie (definitiv)
An eine autoimmune Myositis ist bei Patienten mit einer proximalen Muskelschwäche mit oder ohne Druckschmerzhaftigkeit der Muskulatur zu denken. Dermatomyositis should be suspected in patients with symptoms of myositis and skin findings compatible with dermatomyositis.mit verdächtigen Hautbefunden zu vermuten. Die Diagnosesicherung von Autoimmunmyositis erfordert möglichst viele der folgenden 5 Kriterien:
proximale Muskelschwäche
charakteristischer Hautausschlag
Erhöhte Muskelenzyme im Serum (wenn die Kreatinkinase [CK] nicht erhöht ist, Aminotransferasen oder Aldolase, die weit weniger spezifisch sind als die CK)
charakteristische Muskelveränderungen im EMG oder MRT
muskelbioptische Veränderungen (als definitiver Beweis)
Die Biopsie-Befunde können variieren, typisch ist jedoch eine chronische Entzündung mit Muskeldegeneration und einer gewissen Regeneration. Polymyositis und Dermatomyositis lassen sich häufig durch eine Muskelbiopsie unterscheiden. Vor der Behandlung der Polymyositis wird eine eindeutige Diagnose durch eine Muskelbiopsie empfohlen, um andere Muskelerkrankungen auszuschließen, z. B. solche, die auf fehlende oder defekte Enzyme, nekrotisierende Myositis und postvirale Rhabdomyolyse zurückzuführen sind. Eine Muskelbiopsie ist normalerweise nicht notwendig, wenn Hautbefunde für eine Dermatomyositis charakteristisch sind. Es gibt keinen pathognomonischen Hautbefund für Dermatomyositis bei der Biopsie, aber das Fehlen einer direkten Immunfluoreszenz hilft, den Ausschlag von dem Ausschlag bei Patienten mit systemischem Lupus erythematodes zu unterscheiden.
Um die Sensitivität der Biopsiebefunde zu erhöhen, sollte die Biopsieprobe von einem Muskel stammen, der eine oder mehrere der folgenden Merkmale aufweist:
Schwäche bei klinischer Untersuchung
Muskelödem im MRT identifiziert
im EMG sich als anomal erweisendes kontralaterales Paar eines Muskels
Laboruntersuchungen können den Verdacht auf diese Diagnose erhärten oder schwächen, den Schweregrad bemessen, überlappende Syndrome feststellen und bei der Feststellung von Komplikationen helfen. Auf Autoantikörper sollte untersucht werden. Antinukleäre Antikörper (ANA) sind bei bis zu 80% der Patienten mit Dermatomyositis und Polymyositis positiv. Wenn der ANA-Test positiv ist, sind weitere Tests auf spezifische Antikörperarten wichtig, um den Verdacht auf ein Overlap-Syndrom zu erhöhen.
Der klinische Verlauf und die Manifestationen sind mit bestimmten Antikörpern assoziiert, wie in Tabelle Autoantikörper bei Autoimmun-Myositis. Die Beziehung zwischen diesen Antikörpern und der Pathogenese der Krankheit bleibt unklar, wenngleich anti-Jo-1-Antikörper einen signifikanten Marker für fibrosierende Alveolitis, pulmonale Fibrose, Arthritis und Raynaud-Syndrom darstellen. Es gibt keine für Polymyositis spezifischen Antikörper.
Ein Nachweis für ein erhöhtes Krebsrisiko ist relativ bei Dermatomyositis ausgeprät und bei Polymyositis weniger stark. Daher sollte die Krebsvorsorge für Patienten ≥ 40 Jahre mit Dermatomyositis oder für Patienten ≥ 60 Jahre mit Polymyositis in Betracht gezogen werden, da diese Patienten oft unerwartete Krebserkrankungen haben. Das Screening sollte mindestens eine körperliche Untersuchung umfassen, die Brust, Becken und Rektum (mit okkultem Bluttest), komplettes Blutbild, biochemisches Profil, Mammographie, Urinanalyse, Thoraxröntgen und alle anderen, dem Alter des Patienten entsprechenden Tests umfasst.
Weitere Untersuchungen sollten sich auf die Anamnese und die Befunde der körperlichen Untersuchung stützen. Einige Experten empfehlen ein CT von Thorax, Abdomen und Becken sowie die Darmspiegelung, insbesondere bei Patienten mit Dermatomyositis. Bei jüngeren Patienten ohne Hinweis auf eine Krebserkrankung kann dieses Screening jedoch unterbleiben.
Prognose für Autoimmunmyositis
Langanhaltende Remissionen (oder sogar völliges Abklingen) kommen bei ca. 50% der behandelten Patienten innerhalb von 5 Jahren vor, bei Kindern noch häufiger. Ein Rückfall ist jedoch jederzeit möglich. Alles in allem liegt die 5-Jahres-Überlebensrate bei 75%, bei Kindern darüber.
Zum Tod führen bei Erwachsenen schwere und progrediente Muskelschwäche, Dysphagie, Unterernährung, Aspirationspneumonie oder respiratorisches Versagen mit pulmonaler Superinfektion.
Bei Kindern mit Dermatomyositis kann eine Vaskulitis des Darmes die Todesursache sein.
Dermatomyositis und Polymyositis wurden mit einem erhöhten Krebsrisiko in Verbindung gebracht. Eine Krebserkrankung bestimmt, falls vorhanden, die Gesamtprognose.
Behandlung der Autoimmunmyositis
Kortikosteroide
Immunsuppressiva (z. B. Methotrexat, Azathioprin, Mycophenolatmofetil, Rituximab, Tacrolimus)
Intravenös Immunglobulin
Die körperliche Aktivität sollte bis zum Abklingen der akuten Entzündung mäßig reduziert werden.
Die Initialtherapie besteht aus Kortikosteroiden, Bei akuten Erkrankungen erhalten Erwachsene einmal täglich Prednison 1 mg/kg (in der Regel etwa 40 bis 60 mg) p.o. Bei schweren Erkrankungen mit Dysphagie oder Atemmuskelschwäche beginnt die Behandlung oft mit hochdosierter Kortikosteroidtherapie (z. B. Methylprednisolon 0,5 bis 1g IV einmal täglich für 3 bis 5 Tage).
Serienmessungen von Creatin-Kinase liefern den besten Frühindikator für die therapeutische Wirksamkeit. Bei Patienten mit ausgeprägter Muskelatrophie fällt die Bestimmung jedoch gelegentlich trotz chronischer aktiver Myositis normal aus. MRT-Befunde von Muskelödemen oder hohe CK-Werte unterscheiden im Allgemeinen einen Rückfall der Myositis von einer kortikosteroidinduzierten Myopathie. Aldolase ist eine Alternative, die weniger spezifisch für Muskelverletzungen ist als Kreatinkinase (CK), aber bei Patienten mit Myositis und normalen CK-Spiegeln gelegentlich positiv sein kann. Da die Enzymwerte bei vielen Patienten innerhalb von 6–12 Wochen auf den Normalwert sinken oder diesen erreichen und sich später die Muskelkraft verbessert, kann die Kortikosteroiddosis schrittweise verringert werden. Wenn die Muskelenzymwerte wieder ansteigen, wird die Kortikosteroiddosis in der Regel erhöht, während die volle Wirkung der anderen Medikamente abgewartet wird.
Das Hauptziel besteht darin, die Entzündung schnell zu beseitigen, aber die langfristige Kortikosteroid-Belastung zu minimieren. Deshalb wird ein zweites Medikament (in der Regel Methotrexat, Tacrolimus oder Azathioprin als Nicht-Kortikosteroid-Medikamente der ersten Wahl) gleichzeitig mit den Kortikosteroiden oder kurz danach begonnen, so dass Prednison auf eine Höchstdosis von 5 mg pro Tag reduziert werden kann, idealerweise innerhalb von etwa 6 Monaten. IV-Immunglobulin ist eine gute Option für Patienten, die nicht schnell auf die Therapie ansprechen, die infektiöse Komplikationen mit hochdosierten Kortikosteroiden und anderen Immunsuppressiva entwickeln oder sich einer Chemotherapie unterziehen. Einige Experten wenden eventuell in schweren Fällen oder bei Vorliegen einer Kortikosteroidtoxizität eine Kombination aller 3 Therapien an. Bei Kindern sind Initialdosen von 30–60 mg/m2 einmal täglich erforderlich.
Gelegentlich werden Patienten, die chronisch mit hochdosierten Kortikosteroiden behandelt werden, nach der ersten Reaktion aufgrund einer überlagerten, schmerzfreien Kortikosteroid-Myopathie zunehmend schwach. Bei diesen Patienten bleibt CK normal, obwohl die Patienten schwächer sind.
Myositiden in Assoziation mit einer Krebserkrankung ist refraktär zu Kortikostroiden Die krebsassoziierte Myositis kann abklingen, wenn der Primärtumor entfernt ist.
Menschen mit einer Autoimmunerkrankung haben ein höheres Risiko für Atherosklerose und sollten engmaschig überwacht werden. Patienten mit langfristiger Kortikosteroidtherapie sollten eine Osteoporoseprophylaxe erhalten. Wenn eine immunsuppressive Kombinationstherapie eingesetzt wird, sollten die Patienten eine Prophylaxe für opportunistische Infektionen, wie Pneumocystis jirovecii (siehe Prävention von Pneumocystis jirovecii-Pneumonie), und Impfstoffe gegen häufige Infektionen (z. B. Streptokokkenpneumonie, Influenza, COVID-19) erhalten.
Wichtige Punkte
Die durch Myositis verursachte Muskelschwäche ist meist proximal.
Heliotropischer Hautausschlag und Gottron-Papeln sind spezifisch für Dermatomyositis.
Um die Diagnose zu sichern, ist auf charakteristischen Hautausschlag, Muskelschwäche, erhöhte Creatin-Kinase-Spiegel sowie Muskelveränderungen im EMG oder MRT zu achten.
Es sei denn, die Patienten haben die charakteristischen Hautbefunde, dann machen Sie eine Muskelbiopsie, um die Diagnose zu bestätigen.
Betrachten Sie Screening-Patienten ≥ 40 Jahre mit Dermatomyositis und Patienten ≥ 60 Jahre mit Polymyositis für Krebs.
Behandeln Sie Patienten mit Kortikosteroiden und anderen Immunsuppressiva.