



Tay-Sachs disease and Sandhoff disease are sphingolipidoses, inherited disorders of metabolism, caused by hexosaminidase deficiency that causes severe neurologic symptoms and early death.
Gangliosides are complex sphingolipids present in the brain. There are 2 major forms, GM1 and GM2, both of which may be involved in lysosomal storage disorders. There are 2 main types of GM2 gangliosidosis, each of which can be caused by numerous different mutations.
For more information, see table .
See also Approach to the Patient With a Suspected Inherited Disorder of Metabolism.
Tay-Sachs Disease
Deficiency of hexosaminidase A results in accumulation of GM2 in the brain. Inheritance is autosomal recessive; the most common mutations are carried by 1/27 unaffected adults of Eastern European (Ashkenazi) Jewish origin, although other mutations cluster in some French-Canadian and Cajun populations. The disease develops in 25% of the children when both parents are carriers.
Children with Tay-Sachs disease start missing developmental milestones after age 6 months and develop progressive cognitive and motor deterioration resulting in seizures, intellectual disability, paralysis, and death by age 5 years.
A cherry-red macular spot is common.

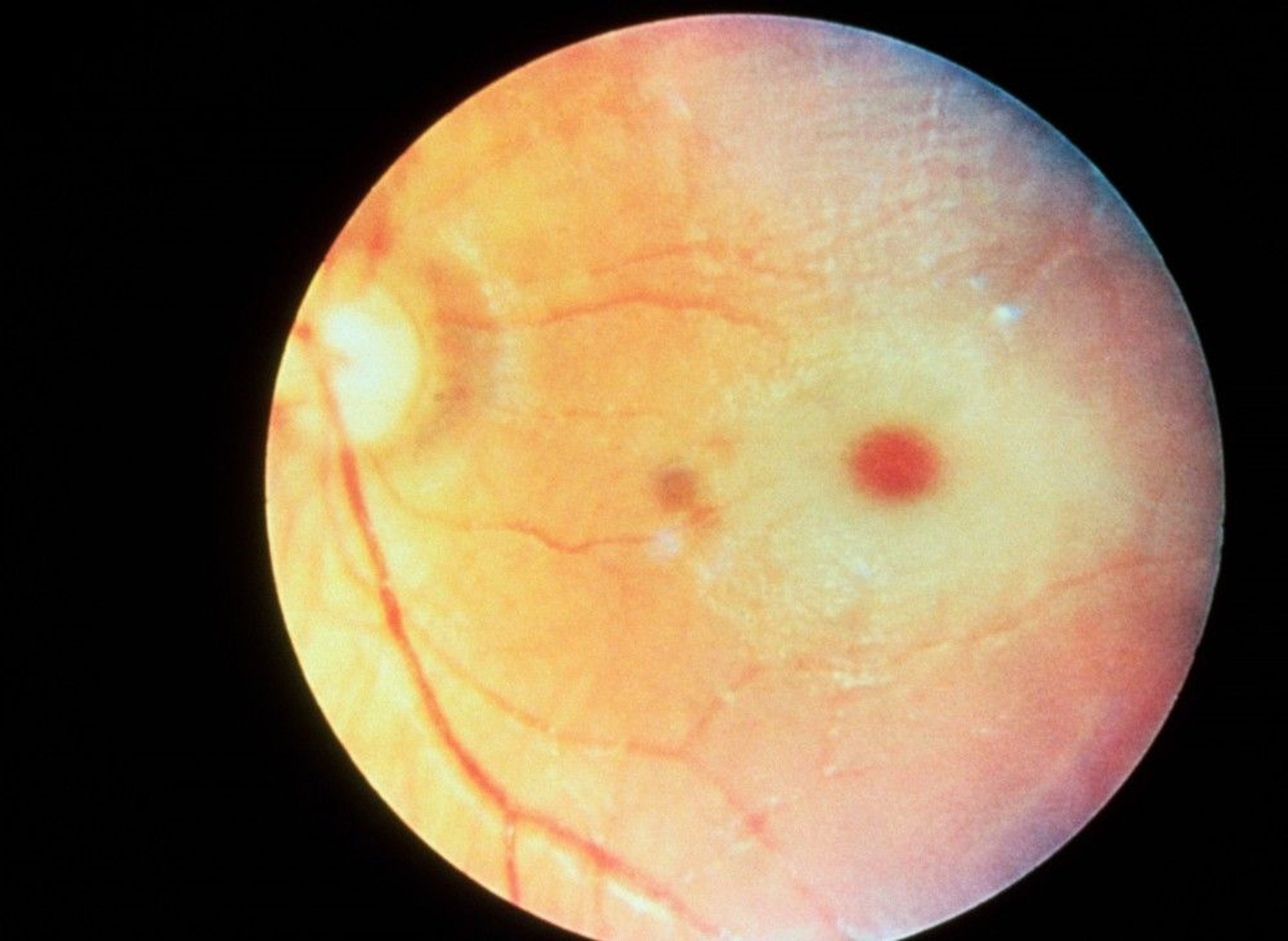
Many patients with Tay-Sachs disease or Sandhoff disease have a cherry-red spot, easily observable by a physician using an ophthalmoscope, in the back of their eye.
RALPH C. EAGLE, JR./SCIENCE PHOTO LIBRARY
Diagnosis of Tay-Sachs disease is clinical and can be confirmed by DNA analysis and/or enzyme assay. (See also testing for suspected inherited disorders of metabolism.)
In the absence of effective treatment, management is focused on screening adults of childbearing age in high-risk populations to identify carriers (by way of enzyme activity and mutation testing) combined with genetic counseling.
Sandhoff Disease
There is a combined hexosaminidase A and B deficiency. Clinical manifestations include progressive cerebral degeneration beginning at 6 months, accompanied by blindness, cherry-red macular spot, and hyperacusis.
Sandhoff disease is almost indistinguishable from Tay-Sachs disease in course, diagnosis, and management, except that there is visceral involvement (hepatomegaly and bone change) and no ethnic association.
More Information
The following English-language resource may be useful. Please note that THE MANUAL is not responsible for the content of this resource.
Online Mendelian Inheritance in Man (OMIM) database: Complete gene, molecular, and chromosomal location information





