




La amiloidosis incluye un grupo de trastornos dispares caracterizados por el depósito extracelular de fibrillas insolubles compuestas por proteínas agrupadas irregularmente. Estas proteínas pueden acumularse en un área y provocar relativamente pocos síntomas o comprometer varios órganos y causar insuficiencia multiorgánica grave. La amiloidosis puede presentarse de novo o ser secundaria a varias infecciones, trastornos inflamatorios o enfermedades malignas. El diagnóstico se realiza mediante una biopsia del tejido afectado; la proteína amiloidogénica se tipifica usando una variedad de técnicas inmunohistológicas y bioquímicas. El tratamiento depende del tipo de amiloidosis.
Las fibrillas de amiloide se componen de proteínas mal plegadas con solubilidad normal que se agregan en oligómeros y luego se convierten en fibrillas insolubles. Una serie de proteínas normales (de tipo salvaje) y mutantes son susceptibles de presentar ese plegamiento y agregación anormales (proteínas amiloidogénicas), lo que explica la gran variedad de causas y tipos de amiloidosis.
Los depósitos de amiloide están formados por pequeñas fibrillas insolubles (de alrededor de 10 nm de diámetro) que forman láminas-beta plegadas congófilas y pueden identificarse por difracción de rayos X. Además de la proteína amiloidea fibrilar, los depósitos también contienen el componente amiloideo P y glucosaminoglucanos.
Los depósitos de amiloide se tiñen de rosa con hematoxilina y eosina, contienen constituyentes de hidratos de carbono que se tiñen con ácido peryódico Schiff o con azul Alcián, pero lo más característico es su birrefringencia verde manzana con microscopia de luz polarizada después de la tinción con rojo Congo. En la autopsia, los órganos afectados pueden tener un aspecto céreo a la inspección.
Para desarrollar amiloidosis, además de la producción de las proteínas amiloidogénicas, es probable que también exista un fracaso de los mecanismos de eliminación normales para este tipo de proteínas mal plegadas. Los depósitos de amiloide en sí mismos son metabólicamente inertes, pero interfieren físicamente con la estructura y el funcionamiento de los órganos. Sin embargo, algunos oligómeros prefibrilares de proteínas amiloidogénicas tienen toxicidad celular directa, un componente importante de la patogenia de la enfermedad.
Etiología de la amiloidosis
En la amiloidosis sistémica, las proteínas amiloidogénicas circulantes forman depósitos en una variedad de órganos. Los principales tipos sistémicos son
AL (amiloidosis primaria): causada por la sobreexpresión adquirida de cadenas ligeras de inmunoglobulina clonal
AF (amiloidosis familiar): causada por la herencia de un gen mutante que codifica una proteína con propensión al mal plegamiento, más comúnmente la transtiretina (TTR)
ATTRwt (amiloidosis sistémica de tipo salvaje; antes conocida como amiloidosis sistemica senil o ASS): causada por un mal plegamiento y agregación del tipo salvaje de transtiretina (TTR).
AA (amiloidosis secundaria): causada por la agregación de un reactante de fase aguda, el amiloide A sérico
Amiloidosis causada por la agregación de beta-2-microglobulina puede ocurrir en pacientes en hemodiálisis por tiempo prolongado, pero la incidencia ha disminuido con el uso de membranas de diálisis modernas de alto flujo. Hay una forma hereditaria rara de amiloidosis beta-2-microglobulina debida a una mutación en el gen relevante.
Las formas localizadas de amiloidosis parecen ser causadas por la producción local y el depósito de una proteína amiloidogénica (con mayor frecuencia inmunoglobulinas de cadena ligera) en el órgano afectado en lugar del depósito de proteínas circulantes. Los sitios más frecuentemente implicados incluyen el sistema nervioso central (p. ej., en la enfermedad de Alzheimer), piel, vías aéreas superiores o inferiores, parénquima pulmonar, vejiga, ojos y mamas.
Amiloidosis AL (amiloidosis primaria)
La amiloidosis primaria es causada por la hiperproducción de una cadena ligera de inmunoglobulina amiloidogénica en pacientes con una célula plasmática monoclonal u otro trastorno linfoproliferativo de células B. Las cadenas ligeras también pueden formar depósitos tisulares no fibrilares (es decir, enfermedad por depósito de cadenas ligeras). En raras ocasiones, las cadenas pesadas de inmunoglobulina forman fibrillas amiloides (denominada amiloidosis AH).
Los sitios más frecuentes donde se deposita el amiloide son la piel, los nervios, el corazón, el tubo digestivo (incluso la lengua), los riñones, el hígado, el bazo y los vasos sanguíneos. Generalmente hay una plasmocitosis leve en la médula ósea, similar a lo observado en un mieloma múltiple, aunque la mayoría de los pacientes no tienen un verdadero mieloma mútilple (con lesiones óseas líticas, hipercalcemia, cilindros tubulares en los riñones y anemia). No obstante, entre el 10 y el 20% de los pacientes con mieloma múltiple también desarrolla amiloidosis AL.
Amiloidosis AF (amiloidosis familiar)
La amiloidosis familiar es causada por la herencia de un gen que codifica una proteína sérica mutada propensa a la agregación, por lo general una proteína producida en abundancia por el hígado.
Las proteínas séricas que pueden causar amiloidosis familiar incluyen la transtiretina (TTR), la apolipoproteína A-I, y A-II, la lisozima, el fibrinógeno, la gelsolina y la cistatina C. Una forma que se especula que es familiar es causada por la proteína del suero factor quimiotáctico de leucocitos 2 (LECT2); sin embargo, no se ha demostrado claramente una mutación genética específica heredada para este último tipo.
La amiloidosis causada por TTR (ATTR) es el tipo más común de amiloidosis familiar. Más de 130 mutaciones del gen de la TTR (transtiretina) se han asociado con amiloidosis. La mutación más frecuente, V30M, es común en Portugal, Suecia, Brasil y Japón, y una mutación V122I está presente en aproximadamente el 4% de los afroestadounidenses y caribeños. La penetrancia de la enfermedad y su edad de aparición son muy variables, pero son constantes dentro de las familias y los grupos étnicos (1).
La ATTR causa neuropatía sensitivomotora periférica y neuropatía autónoma, enfermedad renal crónica y miocardiopatía. El síndrome del túnel carpiano comúnmente precede a otras manifestaciones de enfermedades neurológicas. Los depósitos vítreos pueden desarrollarse debido a la producción de TTR mutante por el epitelio retiniano, o pueden acumularse depósitos leptomeníngeos somo el plexo coroideo produce TTR mutante. Cuando la miocardiopatía es la manifestación predominante del depósito de TTR en el corazón, se denomina miocardiopatía amiloide transtiretina (ATTR-CM).
ATRwt amiloidosis (amiloidosis sistémica senil)
La amiloidosis ATTRwt es causada por la agregación y depósito de TTR de tipo salvaje, con manifestación clínica principalmente en el corazón.
La amiloidosis ATTRwt se reconoce cada vez más como una causa de miocardiopatía infiltrativa en hombres mayores. Aproximadamente el 16% de los pacientes con estenosis aórtica sometidos a un reemplazo de la válvula aórtica transcatéter (2) y el 13% de los hospitalizados por insuficiencia cardíaca con fracción de eyección preservada (HFpEF) también tienen miocardiopatía amiloide transtiretina, en este caso designada como wATTR-CM para indicar el depósito de TTR de tipo salvaje en el corazón (3). Las manifestaciones del amiloide ATTRwt en los tejidos blandos, incluido el síndrome del túnel carpiano, la rotura del tendón bicipital, los desgarros del manguito de los rotadores y la estenosis espinal, pueden preceder en varios años a la expresión clínica de la miocardiopatía infiltrante.
Los factores genéticos y epigenéticos que conducen a la amiloidosis ATTRwt son desconocidos. Debido a que tanto la amiloidosis ATTRwt como la amiloidosis primaria pueden causar miocardiopatía, y porque en los pacientes de este grupo etario pueden haber gammapatías monoclonales amiloidogénicas, es esencial establecer con precisión el tipo de amiloide para que los pacientes con amiloidosis ATTRwt no sean tratados inadecuadamente con quimioterapia (que se utiliza para la amilodosis primaria).
Amiloidosis AA (amiloidosis secundaria)
Esta forma puede ser secundaria a varias infecciones, trastornos inflamatorios y enfermedades malignas y se produce como resultado de la agregación de isoformas del reactante de fase aguda sérico amiloide A.
Las infecciones que causan la enfermedad con mayor frecuencia son
Los trastornos inflamatorios predisponentes incluyen
Síndromes hereditarios de fiebre periódica como fiebre mediterránea familiar
Enfermedad de Castleman
Las citocinas inflamatorias (p. ej., interleucina [IL] -1, factor de necrosis tumoral [TNF], IL-6) sintetizadas en estos trastornos o en forma ectópica por células tumorales causan una producción hepática aumentada del amiloide A sérico (SAA).
La amiloidosis AA secundaria predomina en los riñones, el bazo, el hígado, las glándulas suprarrenales y los ganglios linfáticos. El compromiso del corazón o los nervios periféricos y autónomos ocurre en forma tardía.
Amiloidosis localizada
La amiloidosis localizada fuera del encéfalo es causada con mayor frecuencia por depósitos de cadenas ligeras de inmunoglobulinas clonales; dentro del encéfalo, predomina la proteína beta amiloide.
Los depósitos amiloides localizados en general comprometen las vías respiratorias y el tejido pulmonar, la vejiga y los uréteres, la piel, las mamas, y los ojos. En raras ocasiones, otras proteínas producidas localmente causan la amiloidosis, tales como isoformas de queratina que pueden depositarse en áreas localizadas de la piel. Las cadenas ligeras de inmunoglobulinas clonales producidas por el tejido linfoide asociado a las mucosas en el tubo digestivo, las vías respiratorias, y la vejiga pueden producir amiloidosis primaria en esos órganos.
Los depósitos de proteína amiloide beta en el cerebro contribuyen a la enfermedad de Alzheimer o angiopatía amiloide cerebrovascular. Otras proteínas producidas en el sistema nervioso central pueden plegarse mal, formar agregados y lesionar las neuronas, lo que lleva a enfermedades neurodegenerativas (p. ej., enfermedad de Parkinson, enfermedad de Huntington).
Referencias de la etiología
1. Buxbaum JN, Ruberg FL: Transthyretin V122I (pV142I)* cardiac amyloidosis: an age-dependent autosomal dominant cardiomyopathy too common to be overlooked as a cause of significant heart disease in elderly African Americans. Genet Med 19(7):733-742, 2017. doi:10.1038/gim.2016.200
2. Fabbri G, Serenelli M, Cantone A, et al: Transthyretin amyloidosis in aortic stenosis: clinical and therapeutic implications. Eur Heart J Suppl 23(Suppl E):E128-E132, 2021. doi:10.1093/eurheartj/suab107
3. Magdi M, Mostafa MR, Abusnina W, et al: A systematic review and meta-analysis of the prevalence of transthyretin amyloidosis in heart failure with preserved ejection fraction. Am J Cardiovasc Dis 12(3):102-111, 2022. PMID: 35873185
Síntomas y signos de la amiloidosis
Los síntomas y signos de la amiloidosis sistémica son inespecíficos, lo que a menudo resulta en retrasos en el diagnóstico. La sospecha de amiloidosis debe ser más alta en los pacientes con un proceso de enfermedad multisistémica progresiva.
Los depósitos renales de amiloide que ocurren típicamente en la membrana glomerular producen proteinuria, pero en aproximadamente el 15% de los casos los túbulos se ven afectados, causando azoemia con proteinuria mínima. Estos procesos pueden progresar a síndrome nefrótico con hipoalbuminemia marcada, edema y anasarca o a una nefropatía terminal.
El compromiso hepático genera hepatomegalia indolora, que puede ser masiva. Las pruebas de función hepática por lo general sugieren colestasis intrahepática con elevación de la fosfatasa alcalina y más tarde de la bilirrubina, aunque la ictericia es rara. En ocasiones, se desarrolla hipertensión portal, con várices esofágicas y ascitis.
El compromiso de las vías aéreas y la laringe produce disnea, ronquera, sibilancias, hemoptisis u obstrucción de las vías aéreas.
El infiltrado del miocardio causa una miocardiopatía restrictiva, llevando finalmente a la disfunción diastólica e insuficiencia cardíaca; puede ocurrir bloqueo cardíaco o arritmia. La hipotensión es común.
La neuropatía periférica, con parestesias en los dedos de los pies y las manos, es un signo de presentación frecuente en la amiloidosis primaria y en la ATTR. La neuropatía autónoma puede causar hipotensión ortostática, disfunción eréctil, alteraciones de la sudoración, retención urinaria, y trastornos de la motilidad gastrointestinal.
La angiopatía amiloide cerebrovascular causa con mayor frecuencia una hemorragia cerebral espontánea pero algunos pacientes tienen síntomas neurológicos breves y transitorios.
La amiloidosis gastrointestinal puede causar trastornos de la motilidad del esófago y los intestinos delgado y grueso. También pueden identificarse atonía gástrica, malabsorción, sangrado o seudoobstrucción. En las amiloidosis primarias, la macroglosia es frecuente.
El compromiso por el amiloide de los tejidos blandos precede característicamente a la expresión clínica de la miocardiopatía amiloidea ATTRwt. Las manifestaciones de la enfermedad amiloidea de los tejidos blandos incluyen síndrome del túnel carpiano, dedo en gatillo, rotura del tendón bicipital y estenosis espinal.

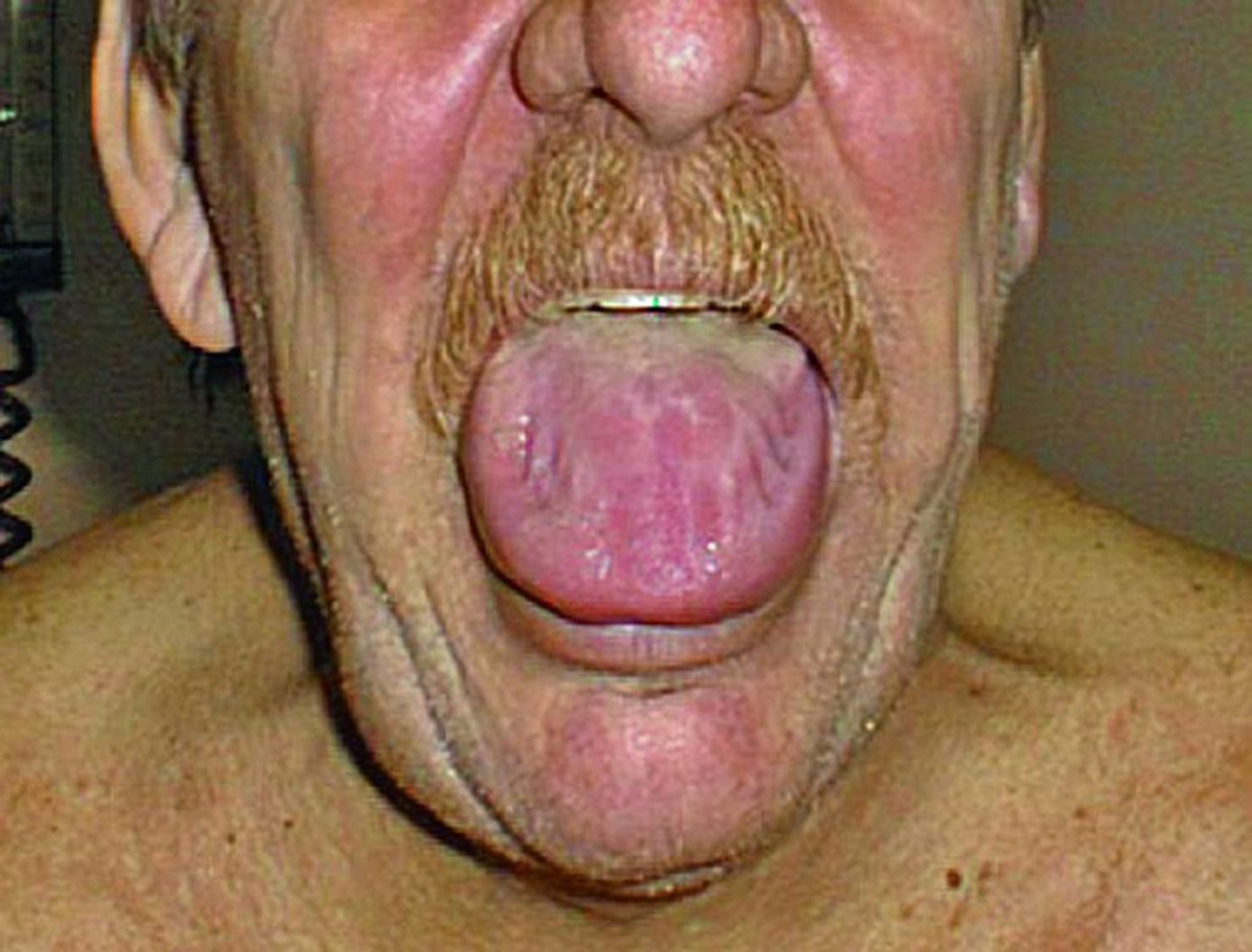
© Springer Science+Business Media
La amiloidosis de la glándula tiroides puede causar un bocio firme, simétrico e indoloro parecido al que se encuentra en la tiroiditis de Hashimoto. También pueden ocurrir otras endocrinopatías.
El compromiso pulmonar (sobre todo en la amiloidosis primaria) puede caracterizarse por nódulos y quistes pulmonares localizados, lesiones traqueobronquiales, derrames pleurales o depósitos alveolares-septales (intersticiales) diseminados.
En varias amiloidosis hereditarias se desrrollan opacidades vítreas amiloideas y márgenes pupilares festoneados bilaterales.
Otras manifestaciones incluyen hematomas, a menudo alrededor de los ojos (ojos de mapache), causados por depósitos de amiloide en los vasos sanguíneos. Los depósitos de amiloide causan debilidad de los vasos sanguíneos, que pueden romperse después de un traumatismo menor, como estornudar o toser.
Diagnóstico de la amiloidosis
Biopsia
Tipo de amiloide
Evaluación del compromiso de los órganos
Biopsia
El diagnóstico de amiloidosis se hace por la demostración de los depósitos fibrilares en un órgano involucrado. La aspiración de grasa abdominal subcutánea detecta depósitos de amiloide en alrededor del 80% de los pacientes con AL, pero menos del 25% de los pacientes con ATTRwt (1). Si el resultado de la biopsia de la grasa es negativo, se debe hacer una biopsia en un órgano comprometido clínicamente. La sensibilidad diagnóstica de las biopsias de riñón y corazón es casi del 100% cuando estos órganos están clínicamente comprometidos. Los cortes tisulares se tiñen con colorante rojo Congo y se examinan con microscopio de luz polarizada para observar la birrefringencia característica. Las fibrillas no ramificadas de 10 nm también pueden ser reconocidas por microscopia electrónica en muestras de biopsia del corazón o los riñones.
La gammagrafía nuclear con marcadores óseos puede diagnosticar miocardiopatía amiloidea por ATTR sin biopsia cardíaca, siempre que se descarte la amiloidosis AL.
Tipo de amiloide
Después de que la amiloidosis ha sido confirmada por biopsia, el tipo se determina usando una variedad de técnicas. Para algunos tipos de amiloidosis, la inmunohistoquímica o la inmunofluorescencia pueden ser diagnósticas, pero hay resultados de tipificación falsos positivos. Otras técnicas útiles incluyen la secuenciación de genes para la AF y la identificación bioquímica por espectrometría de masas para identificar con precisión las variantes de proteínas en los depósitos de amiloide (el método más sensible y específico).
Si se sospecha amiloidosis primaria, los pacientes deben ser evaluados por un trastorno subyacente de las células plasmáticas mediante la medición cuantitativa de cadenas ligeras libres de inmunoglobulina en suero, la detección cualitativa de cadenas ligeras monoclonales en suero u orina utilizando electroforesis de inmunofijación (la electroforesis de proteínas de suero y en orina no es sensible en pacientes con AL), y una biopsia de médula ósea con citometría de flujo o inmunohistoquímica para establecer la clonalidad de las células plasmáticas.
Los pacientes con > 10% de células plasmáticas clonales deben hacerse estudios para ver si cumplen con los criterios para el mieloma múltiple, incluyendo la detección de lesiones óseas líticas, anemia, insuficiencia renal e hipercalcemia.
Compromiso orgánico
Los pacientes son examinados para detectar la afectación de órganos, comenzando con pruebas no invasivas:
Riñones: análisis de orina; medición del nitrógeno ureico en sangre, la creatinina y la albúmina; tasa de filtración glomerular estimada (eTFG); y recolección de orina de 24 horas para la electroforesis de proteínas en orina
Hígado: hepatograma
Pulmones: radiografía de tórax, tomografía computarizada de tórax y pruebas de función pulmonar
Corazón: ECG y concentraciones de biomarcadores como péptido natriurético cerebral (BNP) o N-terminal-pro BNP (NT-pro-BNP) y troponina
El compromiso cardíaco puede ser sugerido por bajo voltaje en las ondas del ECG (causado por un ventrículo engrosado), y/o arritmias. Si se sospecha compromiso cardíaco debido a los síntomas, además de los hallazgos en el ECG y los biomarcadores cardíacos, se realiza una ecocardiografía para medir la relajación diastólica y la tensión longitudinal global (una medida de la función sistólica del ventrículo izquierdo) y para detectar hipertrofia biventricular. En casos ambiguos, se puede realizar una RM cardíaca para detectar el realce de gadolinio subendocárdico persistente, un hallazgo característico. Las exploraciones nucleares cardíacas con pirofosfato de tecnecio han mejorado la detección de la cardiopatía amiloidea ATTR y pueden evitar la necesidad de biopsias cardíacas siempre que los análisis de sangre descarten amiloidosis AL (2, 3).
Referencias del diagnóstico
1. Aimo A, Emdin M, Musetti V, et al: Abdominal Fat Biopsy for the Diagnosis of Cardiac Amyloidosis. JACC Case Rep 2(8):1182-1185, 2020. doi:10.1016/j.jaccas.2020.05.062
2. Gillmore JD, Maurer MS, Falk RH, et al: Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation 133(24):2404–2412, 2016.
3. Maurer MS, Bokhari S, Damy T, et al: Expert consensus recommendations for the suspicion and diagnosis of transthyretin cardiac amyloidosis. Circ Heart Fail 12(9):e006075, 2019.
Tratamiento de la amiloidosis
Tratamiento de sostén
Tratamiento específico
Existen tratamientos específicos para la mayoría de las formas de amiloidosis, aunque algunas terapias están en investigación. Para todas las formas de amiloidosis sistémica, las medidas de soporte pueden ayudar a aliviar los síntomas y mejorar la calidad de vida.
Tratamiento de sostén
Las medidas de soporte están dirigidas al sistema de órganos afectado:
Renal: los pacientes con síndrome nefrótico y edema deben ser tratados con restricción de sal y líquidos y diuréticos de asa; debido a la pérdida continua de proteínas, la ingesta de proteínas no debe restringirse. El trasplante de riñón es una opción cuando se controla el proceso de la enfermedad subyacente, y puede proporcionar supervivencia a largo plazo comparable a la de otras enfermedades renales.
Cardíaca: los pacientes con miocardiopatía deben ser tratados con restricción de sal y líquidos y diuréticos de asa. Otros medicamentos para la insuficiencia cardíaca, incluyendo digoxina, inhibidores de la enzima convertidora de angiotensina (ECA), bloqueantes de los canales de calcio, y beta-bloqueantes, son mal tolerados y están contraindicados. El trasplante cardíaco es exitoso en pacientes muy bien seleccionados con amiloidosis primaria o ATTR y compromiso cardíaco grave. Para prevenir la recurrencia en el corazón trasplantado, los pacientes con amiloidosis AL deben recibir quimioterapia agresiva dirigida al trastorno de células plasmáticas clonales, y los pacientes con polineuropatía amiloidea ATTR sintomática o miocardiopatía deben considerarse candidatos para los tratamientos anti-TTR.
Gastrointestinal: los pacientes con diarrea pueden beneficiarse de la loperamida. Las personas con saciedad precoz y retención gástrica pueden beneficiarse con metoclopramida.
Sistema nervioso: En los pacientes con neuropatía periférica, la gabapentina o la pregabalina o duloxetina pueden aliviar el dolor.
La hipotensión ortostática a menudo mejora con altas dosis de midodrina; este medicamento puede causar retención urinaria en los hombres mayores, pero la complicación de la hipertensión supina rara vez es un problema en esta población. También pueden ser de utilidad las medias de soporte, y se puede usar fludrocortisona en pacientes sin edema periférico, anasarca, o insuficiencia cardíaca. En pacientes con hipotensión ortostática refractaria, puede agregarse midodrina, fludrocortisona o droxidopa.
Amiloidosis primaria (AL)
Para la amiloidosis AL:
La pronta iniciación de la terapia anticélulas plasmáticas es esencial para preservar la función de los órganos y prolongar la vida.
La mayoría de los fármacos utilizados para el mieloma múltiple se han usado en la amiloidosis primaria; la elección del fármaco, la dosis y el esquema de administración con frecuencia deben ser modificados cuando la función del órgano se deteriora.
La quimioterapia usando un agente alquilante (p. ej., melfalán, ciclofosfamida) en combinación con corticosteroides fue el primer régimen en mostrar algún beneficio. El melfalán IV en dosis altas, combinado con trasplante autólogo de células madre puede ser muy eficaz en pacientes seleccionados (1).
Los inhibidores del proteasoma (p. ej., bortezomib) y los inmunomoduladores (p. ej., lenalidomida) también pueden ser eficaces. Un ensayo sobre el anticuerpo monoclonal daratumumab más ciclofosfamida, bortezomib y dexametasona en pacientes con amiloidosis AL recién diagnosticada (que excuyó a los pacientes con insuficiencia cardíaca de clases III y IV de la NYHA, proteína natriurética N-terminal de tipo pro-B [NTproBNP] > 8.500 pg/mL [> 1003 pmol/L], y eGFR < 20 mL/minuto/m2) mostró una alta tasa sin precedentes de respuesta hematológica (2). La respuesta hematológica se basa en la concentración de proteínas monoclonales en suero y orina determinadas con electroforesis de proteínas (inmunofijación) y en las concentraciones séricas de la cadena ligera con los índices kappa/lambda. Sin embargo, no hay datos de supervivencia a largo plazo.
Todos los tratamientos disponibles se dirigen contra las células B clonales o contra las células plasmáticas en la amiloidosis AL. En la actualidad se realizan estudios sobre anticuerpos antifibrillas, como birtamimab y CAEL-101 (3).
La amiloidosis AL primaria puede ser tratada con radioterapia externa en dosis bajas porque las células plasmáticas son altamente sensibles a la radiación.
Amiloidosis ATTR
Para la amiloidosis ATTR:
Trasplante de hígado
Fármacos estabilizadores del tetrámero
Fármacos silenciadores de genes
El trasplante de hígado, que reemplaza el sitio primario de síntesis de la proteína mutante con un nuevo órgano que produce TTR normal, puede ser eficaz en ciertas mutaciones de TTR si se realiza al inicio de la enfermedad (neuropatía temprana y sin compromiso cardíaco). El trasplante que se realiza más adelante en el curso de la enfermedad a menudo conduce al desarrollo de miocardiopatía amiloide progresiva y neuropatía debido al plegamiento anormal y el depósito de la proteína TTR de tipo salvaje en los depósitos de amiloide preexistentes.
Se ha demostrado que varios fármacos estabilizan los tetrámeros de TTR que circulan en el plasma, lo que inhibe el plegamiento anormal de TTR y la formación de fibrillas, y de este modo enlentece de manera eficiente la progresión de la enfermedad neurológica al tiempo que se preserva la calidad de vida. Estos estabilizadores de TTR incluyen diflunisal, un fármaco antiinflamatorio genérico ampliamente disponible y tafamidis (4, 5).
El silenciamiento del gen TTR usando RNA anti-sentido o la interferencia de RNA para bloquear la traducción de TTR mRNA eficientemente reduce eficientemente los niveles séricos de TTR, mejora la evolución neurológica en alrededor del 50% de los pacientes y algunos pacientes parecen capaz de reparar los nervios lesionados en algunos pacientes (6, 7). Se dispone de los medicamentos silenciadores de genes patisiran, inotersen y vutrisiran.
Una prueba de vutrisiran, un silenciador génico de segunda generación, demostró mejores resultados funcionales en pacientes con polineuropatía amiloide familiar (8). Los datos preliminares de otro ensayo sugieren que los silenciadores génicos pueden ser eficaces para el tratamiento de la miocardiopatía en pacientes con amiloidosis ATTR (9).
Amiloidosis ATTRwt
Para la amiloidosis ATTRwt:
Fármacos estabilizadores del tetrámero
Se ha demostrado que la estabilización de la TTR con tafamidis en pacientes con miocardiopatía amiloide ATTR o ATTRwt disminuye la mortalidad por todas las causas y las hospitalizaciones relacionadas con enfermedades cardiovasculares (5). En la actualidad se realizan ensayos clínicos para examinar el efecto de los silenciadores de los genes TTR sobre la miocardiopatía, que se observan en pacientes con amiloidosis ATTRwt, así como en la miocardiopatía que se produce en pacientes con amiloidosis ATTR caracterizada por la proteína mutante (10).
A diferencia de la amiloidosis ATTR hereditaria, el trasplante de hígado no es eficaz en pacientes con ATTRwt porque la proteína amiloidogénica de es una TTR estructuralmente normal.
Amiloidosis secundaria (AA)
Para la amiloidosis AA causada por la fiebre mediterránea familiar, la colchicina por vía oral es eficaz.
Para otros tipos de amiloidosis secundaria, el tratamiento está dirigido a la infección, la enfermedad inflamatoria, o el cáncer.
Se puede usar colchicina o medicamentos anti-IL1, anti-IL6 y anti-TNF para interrumpir la señalización de citocinas, lo que disminuye el proceso inflamatorio que conduce a la producción hepática de amiloide A sérico (AAS).
Referencias del tratamiento
1. Sanchorawala V, Sun F, Quillen K, et al: Long-term outcome of patients with AL amyloidosis treated with high-dose melphalan and stem cell transplantation: 20-year experience. Blood 126: 2345–2347, 2015. doi: 10.1182/blood-2015-08-662726
2. Kastritis E, Palladini G, Minnema MC, et al: Daratumumab-Based Treatment for Immunoglobulin Light-Chain Amyloidosis. N Engl J Med 385(1):46-58, 2021. doi:10.1056/NEJMoa2028631
3. Quarta CC, Fontana M, Damy T, et al: Changing paradigm in the treatment of amyloidosis: From disease-modifying drugs to anti-fibril therapy. Front Cardiovasc Med 9:1073503, 2022. doi:10.3389/fcvm.2022.1073503
4. Berk JL, Suhr OB, Obici L, et al: Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA 310: 2658–2667, 2013. doi: 10.1001/jama.2013.283815
5. Maurer MS, Schwartz JH, Gundapaneni B, et al: Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med 379:1007–1016, 2018.
6. Adams D, Gonzalez-Duarte A, O'Riordan WD, et al: Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med 379:11–21, 2018.
7. Benson MD, Waddington-Cruz M, Berk JL, et al: Inotersen treatment for patients with transthyretin amyloidosis. N Engl J Med 379:22–31, 2018.
8. Adams D, Tournev IL, Taylor MS, et al: Efficacy and safety of vutrisiran for patients with hereditary transthyretin-mediated amyloidosis with polyneuropathy: a randomized clinical trial. Amyloid 30(1):1-9, 2023. doi:10.1080/13506129.2022.2091985
9. Maurer MS, Fontanta MA, Berk JL, et al: Primary results from APOLLO-B, a phase 3 study of patisiran in patients with transthyretin-mediated amyloidosis with cardiomyopathy. Abstract presented at International Symposium of Amyloidosis, September 2022,Heidelberg Germany.
10. Writing Committee, Kittleson MM, Ruberg FL, et al: 2023 ACC Expert Consensus Decision Pathway on Comprehensive Multidisciplinary Care for the Patient With Cardiac Amyloidosis: A Report of the American College of Cardiology Solution Set Oversight Committee [published correction appears in J Am Coll Cardiol 81(11):1135, 2023]. J Am Coll Cardiol 81(11):1076-1126, 2023. doi:10.1016/j.jacc.2022.11.022
Pronóstico de la amiloidosis
El pronóstico depende del tipo de amiloidosis y del órgano comprometido, pero con la atención apropiada y específica para la enfermedad, muchos pacientes tienen una excelente expectativa de vida.
La amilodosis primaria complicada por una miocardiopatía grave tiene peor pronóstico, con una mediana de supervivencia < 1 año. La amiloidosis TTR no tratada generalmente progresa a cardiopatía terminal o enfermedad neurológica dentro de los 5 a 15 años. Se creía que la ATTRwt era la variedad de amiloidosis sistémica que progresaba con mayor lentitud con compromiso del corazón; sin embargo, los pacientes con ATTRwt avanzan a insuficiencia cardíaca sintomática y muerte dentro de una mediana de 4 años desde el diagnóstico de la biopsia.
El pronóstico en la amiloidosis AA depende en gran medida de la eficacia del tratamiento de la enfermedad infecciosa, inflamatoria, o maligna subyacente.
Conceptos clave
La amiloidosis es un grupo de trastornos en los que ciertas proteínas mal plegadas se agregan en fibrillas insolubles que se depositan dentro de los órganos, causando disfunción.
Muchas proteínas diferentes son propensas al mal plegamiento; algunas de estas proteínas son producidas por un defecto genético o por ciertos estados patológicos, mientras que otras son cadenas ligeras de inmunoglobulina producidas por células plasmáticas monoclonales u otros trastornos linfoproliferativos de células B.
La proteína amiloidogénica determina el tipo de amiloide y el curso clínico de la enfermedad, aunque las manifestaciones clínicas de los diferentes tipos pueden superponerse.
Muchos órganos pueden verse afectados, pero la afección cardíaca conlleva un especial mal pronóstico; la miocardiopatía amiloide normalmente conduce a disfunción diastólica, insuficiencia cardíaca, bloqueo cardíaco o arritmia.
El diagnóstico se realiza con biopsia; el tipo de amiloidosis se determina por una variedad de pruebas inmunológicas, genéticas y bioquímicas. La espectrometría de masas es el método más sensible y específico para la tipificación del amiloide.
El tratamiento sintomático apropiado ayudará a aliviar los síntomas y mejorar la calidad de vida; el trasplante de órganos puede ser útil en pacientes seleccionados.
Tratar el proceso subyacente; para la amiloidosis primaria debido a células plasmáticas o trastornos linfoproliferativos, la quimioterapia puede ser muy eficaz; para la amiloidosis secundaria, las terapias antiinfecciosas y los antiinflamatorios pueden ayudar.
Para la amiloidosis ATTR hereditaria, las terapias estabilizadoras de moléculas pequeñas y los fármacos silenciadores de genes inhiben o potencialmente revierten el deterioro neurológico; para los pacientes con miocardiopatía amiloide (ATTR o ATTRwt), el tafamidis disminuye la mortalidad por todas las causas y las hospitalizaciones relacionadas con el aparato cardiovascular.







