




Los gliomas son tumores primarios que se originan en el parénquima encefálico. Los síntomas son diversos, varían según la ubicación, y se manifiestan como déficits neurológicos focales, encefalopatía o convulsiones. El diagnóstico se basa principalmente en la RM, que incluye imágenes stándar ponderadas en T1 y T2, de preferencia con realce de gadolinio, seguida de biopsia con perfil molecular. El tratamiento incluye la resección quirúrgica, la radioterapia y, para algunos tumores, la quimioterapia. La resección pocas veces es curativa.
Los gliomas incluyen
Astrocitomas
Oligodendrogliomas
Glioblastoma multiforme
Ependimomas
Muchos gliomas infiltran el tejido encefálico de forma difusa e irregular.
Los astrorocitomas son los gliomas más frecuentes (véase también Astrocytomas en niños). Se clasifican histológicamente y, en algunos casos, según la presencia de marcadores genéticos específicos, de acuerdo con la clasificación de la OMS (1).
En orden ascendente de malignidad, los astrocitomas se clasifican como
Grado I: astrocitomas pilocíticos y astrocitomas subependimarios de células gigantes (más común en la esclerosis tuberosa)
Grado II: astrocitomas de bajo grado, incluyendo xantoastrocitoma pleomorfo
Grado III: astrocitomas anaplásicos
Grado IV: glioblastomas y gliomas de la línea media difusos
Los astrocitomas pilocíticos, otros de bajo grado o los anaplásicos tienden a desarrollarse en pacientes más jóvenes. Los astrocitomas anaplásicos, en particular, pueden evolucionar más tarde a glioblastomas (llamados glioblastomas secundarios). Los glioblastomas también pueden desarrollarse de novo (llamados glioblastomas primarios), habitualmente en individuos de mediana edad o en personas mayores. Los glioblastomas contienen células cromosómicamente heterogéneas. Tanto los glioblastomas primarios y secundarios tienen características genéticas definidas, que pueden cambiar a medida que los tumores evolucionan. Los glioblastomas secundarios suelen tener mutaciones en los genes IDH1 o IDH2.
En raras ocasiones, los astrocitomas contienen células de astrocitoma y de oligodendroglioma. Estos tumores solían denominarse oligoastrocitomas; sin embargo, ese término ya no se utiliza para referirse a un solo tipo de tumor, sino más bien a una neoplasia mixta.
Oligodendrogliomas (OMS grado II) se encuentran entre los gliomas de crecimiento más lento. Son más frecuentes en el prosencéfalo, sobre todo los lóbulos frontales. Los oligodendrogliomas se caracterizan típicamente por la deleción del brazo p del cromosoma 1 y el brazo q del cromosoma 19 (codeleción 1p/19q). Estas deleciones confirman los tumores oligodendrogliales, predicen una supervivencia más prolongada y una mejor respuesta a la radioterapia y a la quimioterapia. Al igual que los astrocitomas, los oligodendrogliomas pueden evolucionar a formas más agresivas, como los oligodendrogliomas anaplásicos (OMS grado III), que se tratan en forma apropiada.
Tanto los astrocitomas como los oligodendrogliomas pueden expresar mutaciones de los genes IDH1 o IDH2 que conducen a la producción anormal de 2-hidroxiglutarato; este metabolito puede modificar la metilación del DNA de células progenitoras neurales y gliales normales, lo que hace que produzcan células de glioma neoplásicas. Los pacientes con la mutación IDH1/2 tienden a tener un mejor pronóstico que aquellos con tumores IDH1/2 de tipo salvaje, en parte porque los pacientes revelan una mejor respuesta a quimioterapia alquilante, como la temozolomida. Los oligodendrogliomas tienden a tener una codeleción 1p/19q y una mutación IDH1/2. Los astrocitomas suelen tener una mutación en IDH1/2 pero no una codeleción 1p/19q; en cambio, expresan más típicamente mutaciones o pérdida del gen ATRX y mutaciones en pTP53 (2).
Los gliomas de la línea media difusos son tumores astrocíticos de alto grado (grado III a IV de la OMS) que afectan principalmente a los niños. Estos tumores incluyen gliomas pontinos intrínsecos difusos, que son agresivos, típicamente letales ye se infiltran en el tronco encefálico con extensión rostral hacia el hipotálamo y el tálamo y el bulbo raquídeo y la médula espinal en dirección inferior. Los gliomas difusos de la línea media suelen expresar la mutación H3K27M.
Los niños con neurofibromatosis tipo 1 presentan un mayor riesgo de desarrollar gliomas difusos de la línea media.
Los ependimomas ocurren principalmente en niños y adultos jóvenes; son infrecuentes después de la adolescencia (véase también Ependimomas en niños). Se clasifican en
Grado I: subependimoma
Grado II: ependimoma
Grado III: ependimoma anaplásico
Grado IV: ependimoblastoma (que es poco lactantesbebés)
Todos los ependimomas surgen típicamente de la pared ventricular y, por lo tanto, pueden provenir del cerebro, el tronco encefálico o la médula espinal. Como tales, se clasifican según su ubicación: supratentorial, de la fosa posterior o de la médula espinal. Cada una de estas categorías incluye 3 subgrupos definidos molecular e histológicamente, lo que da como resultado 9 tipos de ependimomas con diferencias fenotípicas y moleculares, cuyo tratamiento y pronóstico difieren significativamente entre sí. Los ependimomas del cuarto ventrículo en particular pueden manifestarse con hidrocefalia obstructiva y, por lo tanto, puede manifestarse antes que otros ependimomas (3).
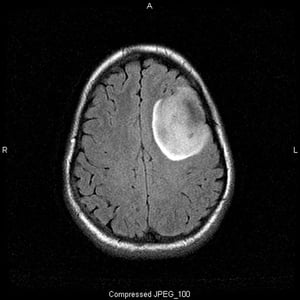
Imagen cortesía de William R. Shapiro, MD.
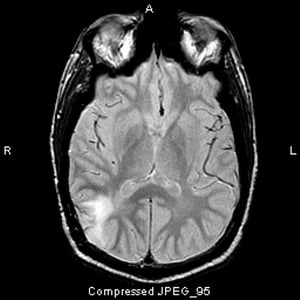
Imagen cortesía de William R. Shapiro, MD.
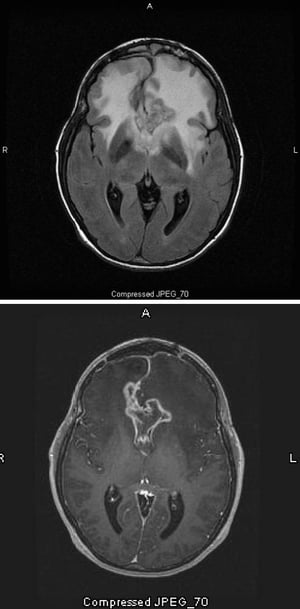
Imágenes cortesía de William R. Shapiro, MD.

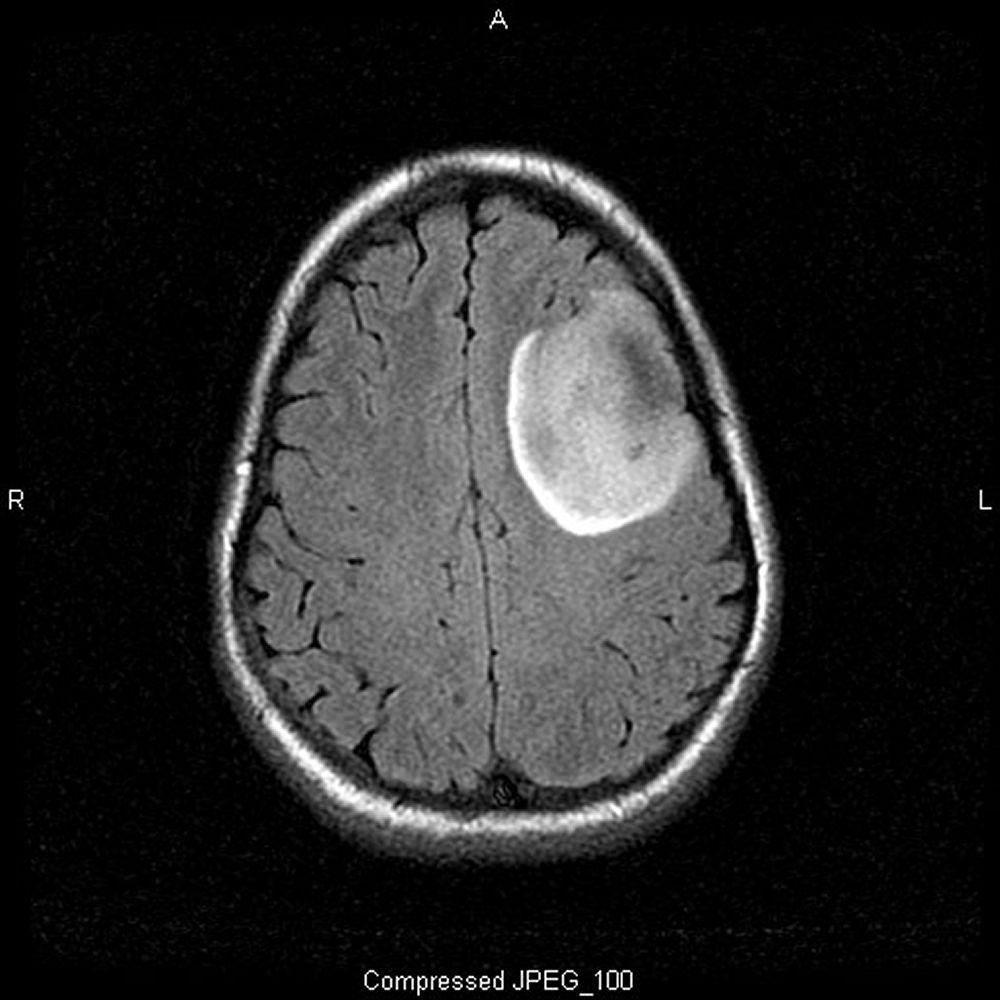
Imagen cortesía de William R. Shapiro, MD.
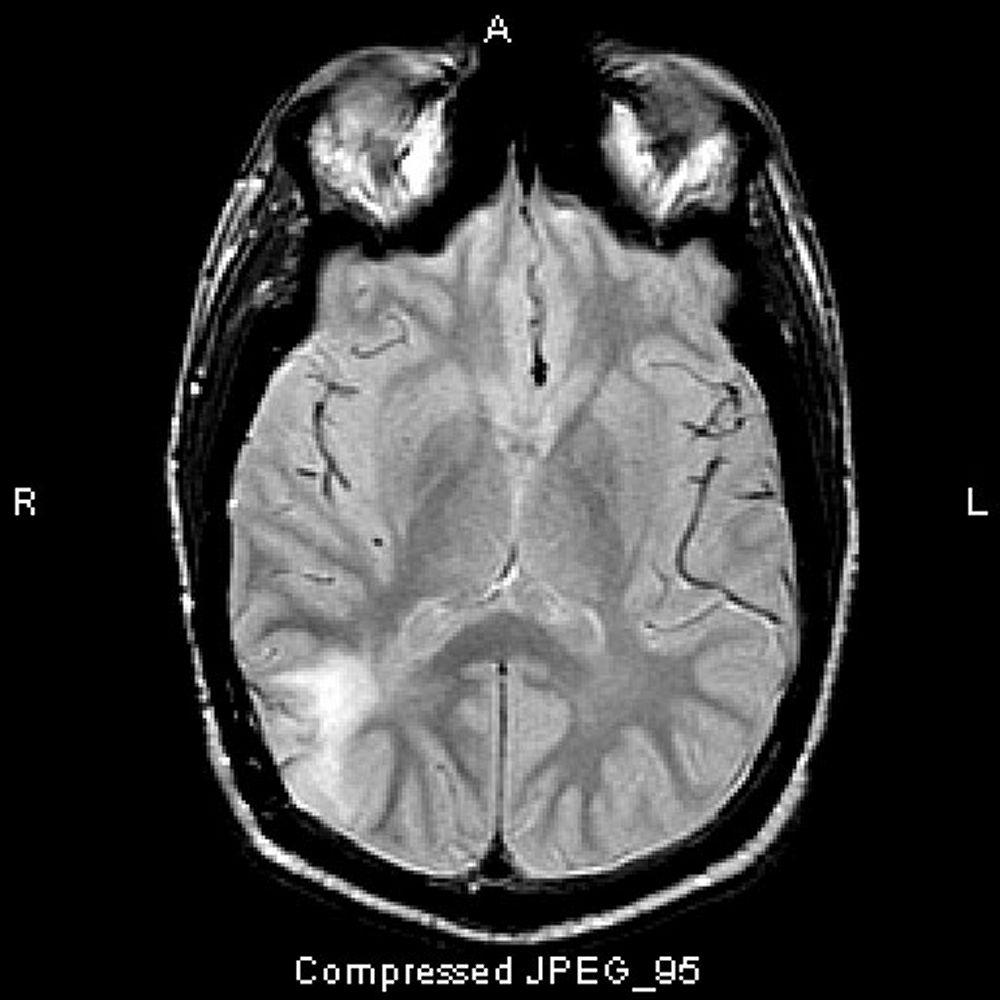
Imagen cortesía de William R. Shapiro, MD.

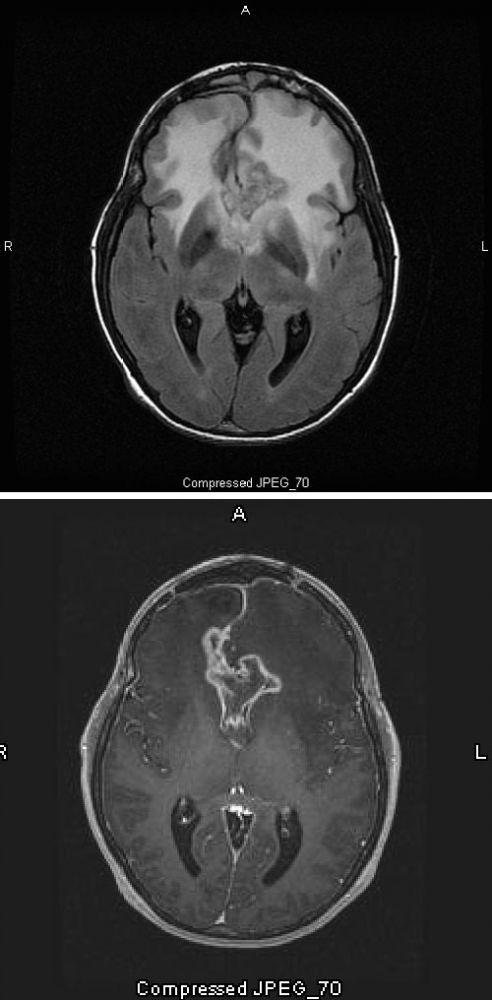
Imágenes cortesía de William R. Shapiro, MD.
Referencias generales
1. Louis DN, Perry A, Reifenberger G, et al: The 2021 World Health Organization classification of tumors of the central nervous system: A summary. Neuro Oncol 23 (8):1231–1251, 2021. doi: 10.1093/neuonc/noab106
2. Reifenberger G, Wirsing H, Knobbe-Thomsen CB, Weller M: Advances in the molecular genetics of gliomas - implications for classification and therapy. Nat Rev Clin Oncol 14 (7):434-452 2017. doi: 10.1038/nrclinonc.2016.204
3. Pajtler K, Mack S, Ramaswamy V, et al: The current consensus on the clinical management of intracranial ependymoma and its distinct molecular variants. Neuropathologica 133:5–12, 2017. doi: 10.1007/s00401-016-1643-0
Diagnóstico de gliomas
RM ponderada en T1 y T2
Biopsia
El diagnóstico de los gliomas se basa principalmente en la RM, que incluye imágenes estándar ponderadas en T1 y T2, de preferencia con realce de gadolinio, seguidas de biopsia con histopatología y perfil molecular. La histopatología incluye el análisis anatomopatológico clásico de la morfología celular, las tinciones inmunohistoquímicas (p. ej., para mutaciones de IDH1/2) e hibridación in situ (p. ej., para mutaciones de EGFR).
Se pueden seleccionar paneles de oncogenes seleccionados, secuenciación dirigida o de exoma completo, secuenciación de RNA y/o análisis de metilación de metilguanina-DNA metil-transferasa (MGMT). La metilación del promotor del gen MGMT es un factor pronóstico en pacientes con glioblastoma multiforme; predice una buena respuesta a la temozolamida posoperatoria y una mayor supervivencia (1).
Referencia del diagnóstico
1. Stupp R, Taillibert S, Kanner A, et al: Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: A randomized clinical trial. JAMA 318 (23):2306–2316, 2017. doi: 10.1001/jama.2017.18718
Tratamiento de gliomas
Resección quirúrgica
Radioterapia
Quimioterapia para algunos tipos
Astrocitomas anaplásicos y glioblastomas
El tratamiento comprende la cirugía, la radioterapia y la quimioterapia para reducir la masa tumoral. La extirpación segura de la mayor cantidad de tumor posible (sin un daño excesivo de áreas importantes del encéfalo [áreas que participan en funciones como el habla y la motricidad]) prolonga la supervivencia y mejora la función neurológica.
Después de la cirugía, los pacientes reciben una dosis tumoral completa de radioterapia (60 Gy en 6 semanas); en condiciones ideales, se utiliza la radioterapia conformada, que se dirige al tumor y respeta el tejido cerebral normal.
Para los glioblastomas, ahora se administra de rutina quimioterapia con temozolomida con la radioterapia. La dosis es de
75/mg/m2/día durante 42 días (incluidos los días de fin de semana cuando no se recibe radiación)
Luego, 150 mg/m2 por vía oral 1 vez al día durante 5 días/mes durante el siguiente mes
Seguido por 200 mg/m2 por vía oral 1 vez al día por 5 días/mes en los meses subsiguientes por un total de 6 a 12 meses
Durante el tratamiento con temozolamida, se administra trimetoprima/sulfametoxazol (TMP/SMX) 800 mg/160 mg 3 veces/semana para prevenir la neumonía por Pneumocystis jirovecii.
Los pacientes que reciben la quimioterapia deben realizar un hemograma a intervalos variados.
En algunos pacientes puede ser apropiado la implantación de reservorios de quimioterapia durante la resección quirúrgica.
En algunos pacientes pueden ser apropiados los campos de tratamiento tumorales más temozolomida adyuvante. Los campos de tratamiento tumoral interfieren con las mitosis del glioblastoma y el ensamblaje de los orgánulos al suministrar campos eléctricos alternos al cuero cabelludo. En un ensayo clínico aleatorizado, los campos de tratamiento tumoral parecieron mejorar la supervivencia en pacientes con glioblastoma (1).
También deben considerarse las terapias experimentales (p. ej., radiocirugía estereotáctica, nuevos agentes quimioterápicos, terapia génica o inmunoterapia). Están en marcha ensayos clínicos con numerosos inhibidores del punto de control inmunitario (checkpoint inhibitors), en particular de la modulación inmunitaria dependiente de CTLA4 y PD1.
Después del tratamiento multimodal convencional, la tasa de supervivencia para los pacientes con glioblastomas es de alrededor del 50% al año, del 25% a los 2 años y del 10 al 15% a los 5 años. El pronóstico es mejor en los siguientes casos:
Los pacientes son < 45 años.
La histología es la de un astrocitoma anaplásico o un tumor de bajo grado (y no la de un glioblastoma).
La resección inicial mejora la función neurológica y deja un tumor residual mínimo o ninguno.
Los tumores tienen la mutación IDH1.
Existe una metilación en el promotor MGMT (metilguanina-metiltransferasa).
Con el tratamiento estándar, el tiempo medio de supervivencia es de aproximadamente 30 meses para los pacientes con astrocitoma anaplásico y unos 15 meses para los pacientes con glioblastoma. Se está realizando una amplia gama de ensayos clínicos, en particular los que incluyen varias inmunoterapias en investigación e inhibidores del punto de control inmunitario (checkpoint inhibitors). Los ensayos en curso y recientemente completados se describen en clinical trials.gov.
Astrocitomas y oligodendrogliomas de bajo grado
La resección quirúrgica segura máxima está indicada para los astrocitomas y los oligodendrogliomas de grado bajo. Después de la resección completa en pacientes < 40 años, se puede considerar la observación. Para otros pacientes, la radioterapia más la quimioterapia adyuvante prolongan la supervivencia (2).
La supervivencia media varía de 1 a 2 años en pacientes de alto riesgo (sin la mutación IDH1, con resección incompleta) a > 10 años en aquellos con factores pronóstico favorables. En pacientes de alto riesgo es probable que la neoplasia maligna progrese más.
Gliomas de la línea media difusos
En pacientes con gliomas difusos de la línea media, radioterapia puede usarse para retrasar la progresión de la enfermedad, aunque su uso es en gran medida paliativo porque la supervivencia suele ser inferior a un año. Estos tumores suelen expresar la mutación H3K27M en una proteína histona; se están estudiando estrategias epigenéticas que modifican el DNA y las proteínas asociadas en la célula para cambiar la expresión génica (3). Los pacientes con la mutación H3K27M tienen mal pronóstico en forma independiente del grado histológico del tumor.
Ependimomas
La punción para analizar el líquido cefalorraquídeo y la imagen craneoespinal deben usarse para estadificar los ependimomas y evaluar la diseminación en el sistema nervioso central.
El tratamiento de los ependimomas incluye la máxima resección quirúrgica segura para la enfermedad unifocal o los tumores sintomáticos. Para los tumores de mayor grado, la radioterapia puede dirigirse localmente o incluir todo el eje craneoespinal en función de la diseminación del tumor. El papel de la quimioterapia no está bien definido.
El pronóstico varía según la localización, la genética y el estadio del tumor (4). En general, con el tratamiento, la tasa global de supervivencia a los 5 años es de un 50%; sin embargo, en los pacientes sin tumor residual, esta tasa a los 5 años es > 70%.
Referencias del tratamiento
1. Stupp R, Taillibert S, Kanner A, et al: Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: A randomized clinical trial. JAMA 318 (23):2306–2316, 2017. doi: 10.1001/jama.2017.18718
2. Buckner JC, Shaw EG, Pugh SL, et al: Radiation plus procarbazine, CCNU, and vincristine in low-grade glioma. N Engl J Med 374 (14):1344-1355, 2016. doi: 10.1056/NEJMoa1500925
3. Miklja Z, Pasternak A, Stallard S, et al: Molecular profiling and targeted therapy in pediatric gliomas: Review and consensus recommendations. Neuro Oncol 21:968–980, 2019.
4. Pajtler K, Mack S, Ramaswamy V, et al: The current consensus on the clinical management of intracranial ependymoma and its distinct molecular variants. Neuropathologica 133:5–12, 2017. doi: 10.1007/s00401-016-1643-0
Conceptos clave
Los gliomas son tumores primarios que se originan en el parénquima encefálico; ellos incluyen los astrocitomas, los oligodendrogliomas y los ependimomas.
Los gliomas varían en ubicación, grado de malignidad, tratamiento y pronóstico.
Para la mayoría de los gliomas, extirpe quirúrgicamente la mayor cantidad de tumor posible (resección total macroscópica) sin dañar áreas del encéfalo importantes, y luego continúe con radioterapia y/o quimioterapia.









