

Acute lymphoblastic leukemia (ALL) is the most common pediatric cancer; it also strikes adults of all ages. Malignant transformation and uncontrolled proliferation of an abnormally differentiated, long-lived hematopoietic progenitor cell results in a high circulating number of blasts, replacement of normal marrow by malignant cells, and the potential for leukemic infiltration of the central nervous system (CNS) and testes. Symptoms include fatigue, pallor, infection, bone pain, CNS symptoms (eg, headache), easy bruising, and bleeding. Examination of peripheral blood smear and bone marrow is usually diagnostic. Treatment typically includes combination chemotherapy to achieve remission, intrathecal and systemic chemotherapy and/or corticosteroids for CNS prophylaxis, and sometimes cerebral irradiation for intracerebral leukemic infiltration, consolidation chemotherapy with or without stem cell transplantation, and maintenance chemotherapy for up to 3 years to avoid relapse.
(See also Overview of Leukemia.)
The American Cancer Society estimates that in the United States in 2023 there will be over 6500 new cases of acute lymphoblastic leukemia (ALL) and almost 1400 deaths will have occurred. Sixty percent of all ALL cases occur in children, with a peak incidence at age 2 to 5 years; a second peak occurs after age 50. ALL is the most common cancer in children, and represents about 75% of leukemias among children < 15 years of age. The risk declines slowly until the mid-20s and then begins to rise again slowly after age 50. ALL accounts for about 20% of adult acute leukemias. The average lifetime risk of ALL in both sexes is about 0.1% (1 in 1000 Americans). Hispanic populations have a higher incidence of ALL than other racial or ethnic populations due in part to polymorphisms in the ARID5B gene.
Pathophysiology of ALL
Similar to acute myeloid leukemia, acute lymphoblastic leukemia is caused by a series of acquired genetic aberrations. Malignant transformation usually occurs at the pluripotent stem cell level, although it sometimes involves a committed stem cell with more limited capacity for self-renewal. Abnormal proliferation, clonal expansion, aberrant differentiation, and diminished apoptosis (programmed cell death) lead to replacement of normal blood elements with malignant cells.
Classification of ALL
In acute lymphoblastic leukemia, the precursor lymphoid neoplasms are broadly categorized based on their lineage into
B-lymphoblastic leukemia/lymphoma (B-ALL/LBL)
T-lymphoblastic leukemia/lymphoma (T-ALL/LBL)
Disease can manifest as a leukemia when neoplastic cells (lymphoblasts) involve blood and bone marrow (defined as > 20% bone marrow blasts) or as a lymphoma when blasts infiltrate mainly extramedullary tissue.
The 2016 World Health Organization (WHO) classification of lymphoid neoplasms incorporates genetic data, clinical features, cell morphology, and immunophenotype, all of which have important implications for disease prognosis and management.
Symptoms and Signs of ALL
Symptoms and signs of acute lymphoblastic leukemia may be present for only days to weeks before diagnosis.
The most common presenting symptoms are due to disrupted hematopoiesis with ensuing
Anemia
Thrombocytopenia
Granulocytopenia
Anemia can manifest with fatigue, weakness, pallor, malaise, dyspnea on exertion, tachycardia, and exertional chest pain.
Thrombocytopenia can cause mucosal bleeding, easy bruising, petechiae/purpura, epistaxis, bleeding gums, and heavy menstrual bleeding. Hematuria and gastrointestinal bleeding are uncommon. Patients can present with spontaneous hemorrhage, including intracranial or intra-abdominal hematomas.
Granulocytopenia or neutropenia can lead to a high risk of infections, including those of bacterial, fungal, and viral etiologies. Patients may present with fevers and a severe and/or recurrent infection.
Organ infiltration by leukemic cells results in enlargement of the liver, spleen, and lymph nodes. Bone marrow and periosteal infiltration may cause bone and joint pain, especially in children with ALL. CNS penetration and meningeal infiltration are common and can result in cranial nerve palsies, headache, visual or auditory symptoms, altered mental status, and transient ischemic attack/stroke.
Diagnosis of ALL
Complete blood count (CBC) and peripheral blood smear
Bone marrow examination
Histochemical studies, cytogenetics, and immunophenotyping
A diagnosis of acute lymphoblastic leukemia is made when blast cells of lymphoid origin are ≥ 20% of marrow nucleated cells or ≥ 20% of non-erythroid cells when the erythroid component is > 50%. If marrow cells are insufficient or unavailable, diagnosis can be made by the same criteria using a peripheral blood sample.

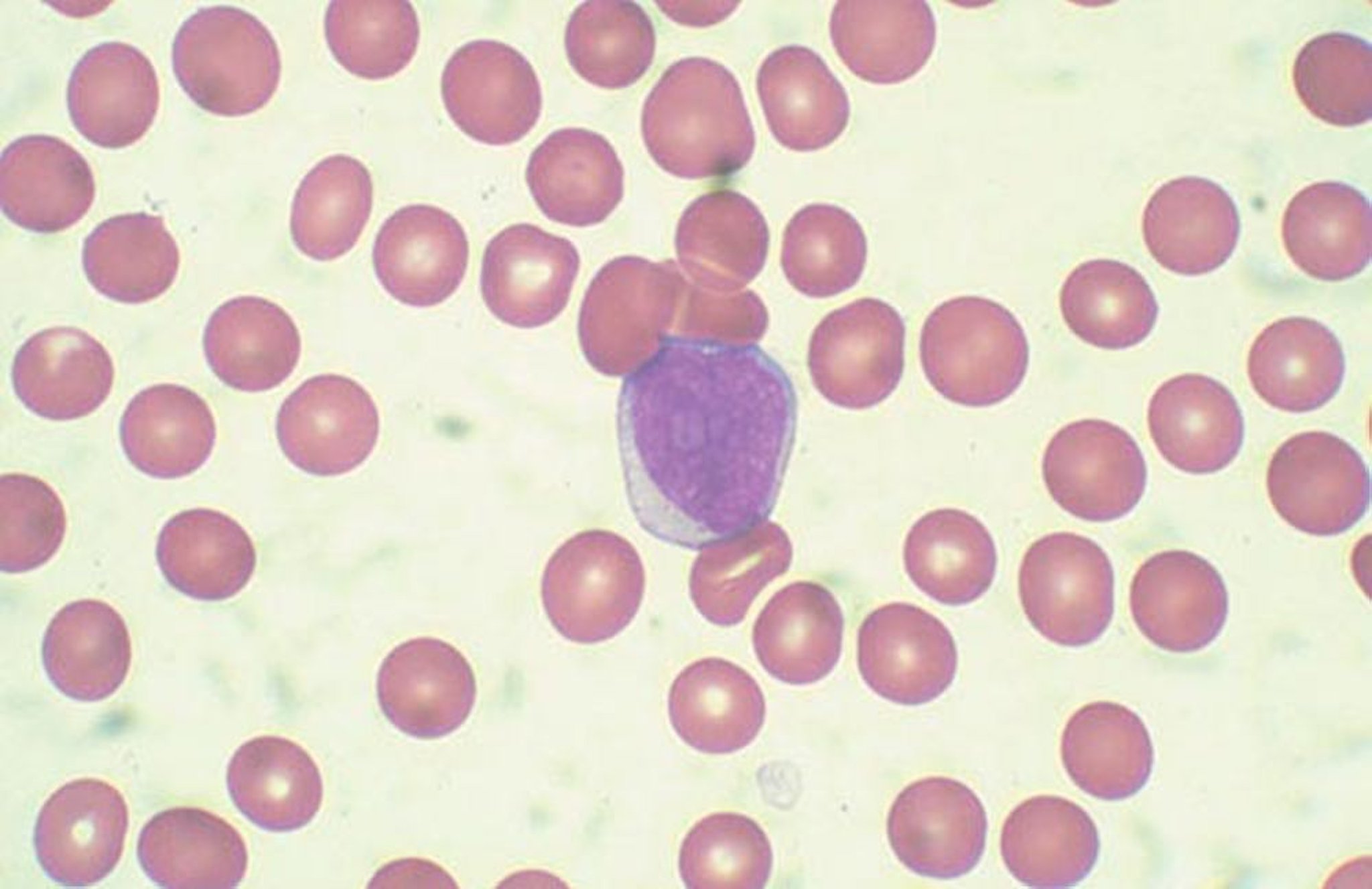
By permission of the publisher. From Chang K, Forman S. In Atlas of Clinical Hematology. Edited by JO Armitage. Philadelphia, Current Medicine, 2004.
CBC and peripheral smear are the first tests done; pancytopenia and peripheral blasts suggest acute leukemia. Blast cells in the peripheral smear may approach 90% of the white blood cell (WBC) count. Aplastic anemia, viral infections such as infectious mononucleosis, and vitamin B12 deficiency, and folate deficiency should be considered in the differential diagnosis of severe pancytopenia. Unlike in AML, Auer rods (linear azurophilic inclusions in the cytoplasm of blast cells) are never present in acute lymphoblastic leukemia.
Bone marrow examination (aspiration and needle biopsy) is routinely done. Blast cells in the bone marrow are typically between 25 and 95% in patients with ALL.
Histochemical studies, cytogenetics, and immunophenotyping studies help distinguish the blasts of ALL from those of AML or other disease processes. Histochemical studies include staining for terminal deoxynucleotidyl transferase (TdT), which is positive in cells of lymphoid origin. Detection of specific immunophenotypic markers such as CD3 (for lymphoid cells of T cell origin) and CD19, CD20, and CD22 (for lymphoid cells of B cell origin) is essential in classifying the acute leukemias. Common cytogenetic abnormalities in ALL include t(9;22) in adults and t(12;21) and high hyperdiploidy in children (see table ).
Common Cytogenetic Abnormalities in ALL
Cytogenetic Abnormality | Estimated Incidence (Adults) | Estimated Incidence (Children) | Prognosis |
|---|---|---|---|
t(9;22)/BCR-ABL1; (Philadelphia chromosome positive or Ph+) | 25% | 5% | Poor |
High hyperdiploidy (51–65 chromosomes in leukemia cells) | 2–11% | 25% | Favorable |
Hypodiploidy (< 46 chromosomes in leukemia cells) | 5% | 5% | Poor |
t(12;21)/TEL-AML1 (ETV6-RUNX1) | < 3% | 20–25% | Favorable |
Data from Cytogenetic abnormalities in adult acute lymphoblastic leukemia: correlations with hematologic findings outcome. A Collaborative Study of the Group Français de Cytogénétique Hématologique [published correction appears in Blood 1996 Oct 1;88(7):2818]. Blood 1996;87(8):3135-3142, Fenaux P, Lai JL, Morel P, et al. Cytogenetics and their prognostic value in childhood and adult acute lymphoblastic leukemia (ALL) excluding L3. Hematol Oncol 1989;7(4):307-317. doi:10.1002/hon.2900070409, Pui CH, Crist WM, Look AT. Biology and clinical significance of cytogenetic abnormalities in childhood acute lymphoblastic leukemia. Blood 1990;76(8):1449-1463, Bloomfield CD, Secker-Walker LM, Goldman AI, et al. Six-year follow-up of the clinical significance of karyotype in acute lymphoblastic leukemia. Cancer Genet Cytogenet 1989;40(2):171-185. doi:10.1016/0165-4608(89)90023-x, Walters R, Kantarjian HM, Keating MJ, et al. The importance of cytogenetic studies in adult acute lymphocytic leukemia. Am J Med 1990;89(5):579-587. doi:10.1016/0002-9343(90)90175-d, and Micallef-Eynaud PD, Eden OB, Grace E, Ellis PM. Cytogenetic abnormalities in childhood acute lymphoblastic leukemia. Pediatr Hematol Oncol 1993;10(1):25-30. doi:10.3109/08880019309016524. | |||
Less common cytogenetic abnormalities include the following:
t(v;11q23) /MLL or KMT2A rearranged, including t(4;11)/KMT2A-AF4
t(1;19)/E2A-PBX1 (TCF3-PBX1)
t(5;14)/IL3-IGH
t(8;14), t(8;22), t(2;8)/C-MYC rearranged
BCR-ABL-like acute lymphoblastic leukemia overlaps phenotypically with ALL in which the Philadelphia chromosome [a reciprocal balanced translocation between chromosomes 9 and 22, t(9;22)] is present (Ph+ ALL).
Other laboratory findings may include hyperuricemia, hyperphosphatemia, hyperkalemia, hypocalcemia, and elevated lactate dehydrogenase (LDH), which indicate a tumor lysis syndrome. Elevated serum levels of hepatic transaminases or creatinine, and hypoglycemia may also be present. Patients with Ph+ ALL and patients with t(v;11q23) involving MLL rearrangements often present with hyperleukocytosis.
CT of the head is done in patients with CNS symptoms. CT of the chest and abdomen should be done to detect mediastinal masses and lymphadenopathy and may also detect hepatosplenomegaly. Echocardiography or multi-gated acquisition (MUGA) scanning is typically done to assess baseline cardiac function (prior to administration of anthracyclines, which are cardiotoxic).
Treatment of ALL
Systemic chemotherapy
Prophylactic CNS chemotherapy and sometimes CNS radiation
For Ph+ ALL, also a tyrosine kinase inhibitor
Supportive care
Sometimes immunotherapy, targeted therapy, stem cell transplantation, and/or radiation therapy
Treatment for newly diagnosed acute lymphoblastic leukemia generally consists of 3 to 4 cycles of chemotherapy blocks of non–cross-resistant chemotherapy for the first 9 to 12 months, followed by 2.5 to 3 years of maintenance chemotherapy.
Chemotherapy
The 4 general phases of chemotherapy for acute lymphoblastic leukemia include
Remission induction
Postremission consolidation
Interim maintenance and intensification
Maintenance
The goal of induction treatment is complete remission, defined as < 5% blast cells in the bone marrow, an absolute neutrophil count > 1000/mcL (> 1 × 109/L), a platelet count > 100,000/mcL (> 100 × 109/L), and no need for blood transfusion. In patients with complete remission, a low measurable residual disease (also known as minimal residual disease or MRD) is the most important prognostic factor (1). Measurable or minimal residual disease is microscopic disease that is not detected by standard assays but can be measured by more sensitive assays. A low measurable residual disease (MRD negativity) is defined variably (based on the assay used) as < 0.01 to 0.1% leukemic cells in bone marrow.
Components of induction therapy include
A high-dose corticosteroid (eg, dexamethasone, prednisone)A high-dose corticosteroid (eg, dexamethasone, prednisone)
An anthracycline (eg, daunorubicin, doxorubicin, idarubicin)An anthracycline (eg, daunorubicin, doxorubicin, idarubicin)
VincristineVincristine
Some regimens use a corticosteroid to reduce disease burden prior to intensive induction. In younger adults, a regimen that includes asparaginase and/or cyclophosphamide for induction, similar to treatment protocols used in children, may increase rates of response and achievement of undetectable minimal residual disease. If complete remission is not achieved after induction, some regimens recommend a second induction course to try to get more patients to complete remission before consolidation. Some regimens use a corticosteroid to reduce disease burden prior to intensive induction. In younger adults, a regimen that includes asparaginase and/or cyclophosphamide for induction, similar to treatment protocols used in children, may increase rates of response and achievement of undetectable minimal residual disease. If complete remission is not achieved after induction, some regimens recommend a second induction course to try to get more patients to complete remission before consolidation.
For patients with Philadelphia chromosome–positive (Ph+) ALL, a tyrosine kinase inhibitor (eg, imatinib, dasatinib) can be added to the drug regimen. For patients with CD20 positive B-lymphoblastic leukemia, rituximab can be added. For patients with Philadelphia chromosome–positive (Ph+) ALL, a tyrosine kinase inhibitor (eg, imatinib, dasatinib) can be added to the drug regimen. For patients with CD20 positive B-lymphoblastic leukemia, rituximab can be added.
The goal of consolidation is to prevent leukemic regrowth. Consolidation therapy usually lasts a few months and combines regimen-specific courses of non–cross-resistant drugs that have different mechanisms of action. For adults with Ph+ ALL, allogeneic stem cell transplantation is recommended as consolidation therapy.
Interim maintenance and late/delayed intensification therapy are used after consolidation therapy. These phases of therapy incorporate a variety of chemotherapeutic agents with different doses and schedules that are less intense than induction and consolidation.
Most regimens include maintenance therapy with monthly vincristine, weekly methotrexate, daily mercaptopurine, and 5 days/month corticosteroid. Therapy duration is usually 2½ to 3 years.with monthly vincristine, weekly methotrexate, daily mercaptopurine, and 5 days/month corticosteroid. Therapy duration is usually 2½ to 3 years.
CNS prophylaxis starts during induction and continues throughout all phases of treatment. Because lymphoblasts often infiltrate the cerebrospinal fluid and meninges, all regimens include CNS prophylaxis and treatment with intrathecal methotrexate, cytarabine, and hydrocortisone in combination or as monotherapy. High doses of systemic methotrexate and/or cytarabine penetrate the CNS, providing extra CNS prophylaxis if regimens include these drugs. Cranial nerve or whole-brain irradiation was previously often done for patients at high risk of CNS disease (eg, high WBC count, high serum lactate dehydrogenase, B-cell phenotype), but its use has been decreasing in recent years.starts during induction and continues throughout all phases of treatment. Because lymphoblasts often infiltrate the cerebrospinal fluid and meninges, all regimens include CNS prophylaxis and treatment with intrathecal methotrexate, cytarabine, and hydrocortisone in combination or as monotherapy. High doses of systemic methotrexate and/or cytarabine penetrate the CNS, providing extra CNS prophylaxis if regimens include these drugs. Cranial nerve or whole-brain irradiation was previously often done for patients at high risk of CNS disease (eg, high WBC count, high serum lactate dehydrogenase, B-cell phenotype), but its use has been decreasing in recent years.
Medically frail patients with ALL
About one third of patients with acute lymphoblastic leukemia are older adults (> 65). Older ALL patients are more likely to have precursor B-cell ALL and have higher risk and more complex cytogenetics, including Philadelphia chromosome positive (Ph+) or t(v;11q23) MLL or KMT2A rearranged disease.
Some, but not all, older patients can tolerate standard induction therapy. Subsequent treatment regimens (CNS prophylaxis, postremission consolidation or intensification, and maintenance) depend on the individual patient's comorbidities and performance status. For example, older patients with several comorbidities and poor performance status may undergo gentler induction therapy without consolidation or maintenance. In older patients with Ph+ ALL, tyrosine kinase inhibitors (eg, imatinib, dasatinib) plus corticosteroids given with either low-intensity or no chemotherapy have resulted in 95 to 100% complete remission rate, with a 45 to 50% 2-year relapse free survival and about 70% 2-year overall survival. For older patients with ALL who are in their first complete remission, nonmyeloablative or reduced-intensity conditioning allogeneic Some, but not all, older patients can tolerate standard induction therapy. Subsequent treatment regimens (CNS prophylaxis, postremission consolidation or intensification, and maintenance) depend on the individual patient's comorbidities and performance status. For example, older patients with several comorbidities and poor performance status may undergo gentler induction therapy without consolidation or maintenance. In older patients with Ph+ ALL, tyrosine kinase inhibitors (eg, imatinib, dasatinib) plus corticosteroids given with either low-intensity or no chemotherapy have resulted in 95 to 100% complete remission rate, with a 45 to 50% 2-year relapse free survival and about 70% 2-year overall survival. For older patients with ALL who are in their first complete remission, nonmyeloablative or reduced-intensity conditioning allogeneichematopoietic stem cell transplantation is an option.
Targeted immunotherapy drugs that are available for treatment of relapsed or refractory ALL are increasingly used for treatment of older patients with ALL in clinical trials or clinical practice.
Older patients with ALL probably tolerate asparaginase more poorly than younger patients do.
Relapsed or refractory ALL
Leukemic cells may reappear in the bone marrow, CNS, testes, or other sites. Bone marrow relapse is particularly ominous. Although a new round of chemotherapy may induce a second remission in the majority of children and about one third of adults, subsequent remissions tend to be brief. Chemotherapy helps only a few patients with early bone marrow relapse to achieve long disease-free second remissions or cure.
New immunotherapy approaches show impressive results in relapsed/refractory ALL. Antibodies, such as blinatumomab, that bring T cells into close proximity to leukemic blasts demonstrate activity in relapsed ALL. New immunotherapy approaches show impressive results in relapsed/refractory ALL. Antibodies, such as blinatumomab, that bring T cells into close proximity to leukemic blasts demonstrate activity in relapsed ALL.Chimeric antigen receptor T (CAR-T) cells, engineered and generated from the patient's T cells, induce remission in patients with relapsed ALL with remarkable efficacy, albeit with significant toxicity (2).
Available immunotherapies for relapsed or refractory ALL include
BlinatumomabBlinatumomab
Inotuzumab ozogamicinInotuzumab ozogamicin
TisagenlecleucelTisagenlecleucel
Blinatumomab,Blinatumomab, a biospecific CD19-directed CD3 T-cell engager, prolongs overall survival for children and adults with relapsed or refractory B-cell precursor ALL, whether Ph+ or Ph-. Life-threatening toxicities may include cytokine release syndrome and neurologic toxicities (eg, seizures, encephalopathy with altered consciousness, and disordered speech, coordination, and/or balance). Interrupting or stopping blinatumomab with or without use of high-dose dexamethasone may be necessary. The most common neurologic symptoms after blinatumomab use are headache and tremor (and neurologic toxicities (eg, seizures, encephalopathy with altered consciousness, and disordered speech, coordination, and/or balance). Interrupting or stopping blinatumomab with or without use of high-dose dexamethasone may be necessary. The most common neurologic symptoms after blinatumomab use are headache and tremor (3).
Inotuzumab ozogamicin,Inotuzumab ozogamicin, a CD22-directed antibody-drug conjugate with calicheamicin, is also available for adults with relapsed or refractory B-cell precursor ALL. One study found significantly higher remission rates after 1 to 2 cycles of therapy with inotuzumab ozogamicin than with standard chemotherapy (a CD22-directed antibody-drug conjugate with calicheamicin, is also available for adults with relapsed or refractory B-cell precursor ALL. One study found significantly higher remission rates after 1 to 2 cycles of therapy with inotuzumab ozogamicin than with standard chemotherapy (4). Inotuzumab may cause hepatotoxicity, including fatal and life-threatening veno-occlusive disease and is associated with higher post-transplant non-relapse mortality.
Tisagenlecleucel,Tisagenlecleucel, a CD19-directed genetically modified autologous T-cell immunotherapy, is available for the treatment of patients up to 25 years of age with B-cell precursor ALL that is refractory or in a 2nd or later relapse. Life-threatening toxicities may include cytokine release syndrome and neurologic toxicities (5).
Brexucabtagene autoleucel, a CD19-directed genetically modified autologous T cell immunotherapy, can be used to treat adult patients with relapsed or refractory B-cell precursor ALL. Complications, including cytokine release syndrome and neurologic toxicities, may be life threatening.
Other agents have been available, but clinically meaningful outcomes have not been convincingly demonstrated (ie, the approvals were based upon response rate but there were no trials verifying an improvement in disease-related symptoms or increased survival) for these. Examples include:
Liposomal vincristine (a vinca alkaloid): For adults with Ph- ALL in at least 2nd relapse or that has progressed despite ≥ 2 antileukemia therapy—marketing has been discontinuedLiposomal vincristine (a vinca alkaloid): For adults with Ph- ALL in at least 2nd relapse or that has progressed despite ≥ 2 antileukemia therapy—marketing has been discontinued
Clofarabine (a purine nucleoside analog): For patients age 1 to 21 years with relapsed or refractory ALL after ≥ 2 prior regimensClofarabine (a purine nucleoside analog): For patients age 1 to 21 years with relapsed or refractory ALL after ≥ 2 prior regimens
Nelarabine (a purine nucleoside) analog prodrug of guanosine arabinoside: For T-cell ALL that has not responded to or has relapsed after ≥ 2 prior regimensNelarabine (a purine nucleoside) analog prodrug of guanosine arabinoside: For T-cell ALL that has not responded to or has relapsed after ≥ 2 prior regimens
Stem cell transplantation following reinduction chemotherapy or immunotherapy offers the greatest hope of long-term remission or cure if an HLA-matched sibling is available. Cells from other relatives or from matched, unrelated donors are sometimes used. Transplantation is rarely used for patients > 65 years because it is much less likely to be successful and because adverse effects are much more likely to be fatal.
CNS relapse treatment includes intrathecal methotrexate (with or without cytarabine or corticosteroids) twice weekly until all signs disappear. The role of continued intrathecal drug use or CNS irradiation is unclear.treatment includes intrathecal methotrexate (with or without cytarabine or corticosteroids) twice weekly until all signs disappear. The role of continued intrathecal drug use or CNS irradiation is unclear.
Testicular relapse may be evidenced clinically by painless firm swelling of a testis or may be identified on biopsy. If unilateral testicular involvement is clinically evident, the apparently uninvolved testis should undergo biopsy. Treatment is radiation therapy of the involved testis and administration of systemic reinduction therapy.
Supportive care
Supportive care is similar in the acute leukemias and may include
Transfusions
Antimicrobials
Hydration and urine alkalinization
Psychologic support
Transfusions of red blood cells and sometimes platelets are administered as needed to patients with bleeding or anemia. Prophylactic platelet transfusion is done when platelets fall to < 10,000/mcL (< 10 × 109/L). Anemia (hemoglobin < 7 or 8 g/dL [< 70 to 80 g/L]) is treated with transfusions of packed red blood cells. Granulocyte transfusions are not routinely used.
Antimicrobials are often needed for prophylaxis and treatment because patients are immunosuppressed; in such patients, infections can progress quickly with little clinical prodrome. After appropriate studies and cultures have been done, febrile patients with neutrophil counts < 500/mcL (< 0.5 × 109/L) should begin treatment with a broad-spectrum bactericidal antibiotic that is effective against gram-positive and gram-negative organisms (eg, ceftazidime, piperacillin/tazobactam, meropenem). Fungal infections, especially pneumonias, may develop and are difficult to diagnose, so chest CT to detect fungal pneumonia should be done early (ie, within 72 hours of presentation with neutropenic fever, depending on the degree of suspicion). Empiric antifungal therapy should be given if antibacterial therapy is not effective within 72 hours. There is a significant drug-drug interaction between vincristine, which is commonly used in all ALL treatment regimens, and azole antifungals. In patients with refractory pneumonitis, /L) should begin treatment with a broad-spectrum bactericidal antibiotic that is effective against gram-positive and gram-negative organisms (eg, ceftazidime, piperacillin/tazobactam, meropenem). Fungal infections, especially pneumonias, may develop and are difficult to diagnose, so chest CT to detect fungal pneumonia should be done early (ie, within 72 hours of presentation with neutropenic fever, depending on the degree of suspicion). Empiric antifungal therapy should be given if antibacterial therapy is not effective within 72 hours. There is a significant drug-drug interaction between vincristine, which is commonly used in all ALL treatment regimens, and azole antifungals. In patients with refractory pneumonitis,Pneumocystis jirovecii infection or a viral infection should be suspected and confirmed by bronchoscopy and bronchoalveolar lavage and treated appropriately.
Posaconazole, a 2nd-generation triazole antifungal drug, is indicated for primary prophylaxis in patients age > 13 years who are at high risk of developing invasive Posaconazole, a 2nd-generation triazole antifungal drug, is indicated for primary prophylaxis in patients age > 13 years who are at high risk of developing invasiveAspergillus and Candida infections because of immunosuppression (eg, hematopoietic stem cell transplant recipients with graft-versus-host disease). In patients with drug-induced immunosuppression (eg, prolonged use of corticosteroids for ALL treatment), sulfamethoxazole/trimethoprim (TMP/SMX), dapsone, atovaquone, or pentamidine is indicated to prevent infections because of immunosuppression (eg, hematopoietic stem cell transplant recipients with graft-versus-host disease). In patients with drug-induced immunosuppression (eg, prolonged use of corticosteroids for ALL treatment), sulfamethoxazole/trimethoprim (TMP/SMX), dapsone, atovaquone, or pentamidine is indicated to preventP. jirovecii pneumonia. Acyclovir or valacyclovir prophylaxis is generally recommended for all patients.pneumonia. Acyclovir or valacyclovir prophylaxis is generally recommended for all patients.
Hydration, urine alkalinization with IV sodium bicarbonate, and allopurinol or rasburicase can prevent and treat the hyperuricemia, hyperphosphatemia, hypocalcemia, and hyperkalemia (ie, urine alkalinization with IV sodium bicarbonate, and allopurinol or rasburicase can prevent and treat the hyperuricemia, hyperphosphatemia, hypocalcemia, and hyperkalemia (ie,tumor lysis syndrome) caused by the rapid lysis of leukemic cells during initial therapy in ALL. Hyperuricemia is minimized by reducing the conversion of xanthine to uric acid by giving allopurinol (a xanthine oxidase inhibitor) or rasburicase (a recombinant urate-oxidase enzyme) before starting chemotherapy. Patients with ) caused by the rapid lysis of leukemic cells during initial therapy in ALL. Hyperuricemia is minimized by reducing the conversion of xanthine to uric acid by giving allopurinol (a xanthine oxidase inhibitor) or rasburicase (a recombinant urate-oxidase enzyme) before starting chemotherapy. Patients withG6PD deficiency may develop severe hemolysis from rasburicase, which is contraindicated in these patients.may develop severe hemolysis from rasburicase, which is contraindicated in these patients.
Psychologic support may help patients and their families with the shock of illness and the rigors of treatment for a potentially life-threatening condition.
Treatment references
1. Berry DA, Zhou S, Higley H, et al: Association of minimal residual disease with clinical outcome in pediatric and adult acute lymphoblastic leukemia: A meta-analysis. JAMA Oncol 3(7): e170580, 2017. doi:10.1001/jamaoncol.2017.0580
2. Lee DW, Kochenderfer JN, Stetler-Stevenson M, et al: T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 385(9967) :517–528, 2015.
3. Kantarjian H, Stein A, Gökbuget N, et al: Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med 376(9):836–847, 2017.
4. Kantarjian HM, DeAngelo DJ, Stelljes M, et al: Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N Engl J Med 375(8):740–753, 2016.
5. Maude SL, Laetsch TW, Buechner J, et al: Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med 378(5):439–448, 2018.
Prognosis for ALL
Prognostic factors help determine treatment protocol and intensity.
Favorable prognostic factors are
Age 3 to 9 years
WBC count < 25,000/mcL (< 25 × 109/L) or < 50,000/mcL (< 50 × 109/L) in children
Leukemic cell karyotype with high hyperdiploidy (51 to 65 chromosomes), t(1;19), and t(12;21)
No CNS disease at diagnosis
Unfavorable factors include
Leukemic cell karyotype with 23 chromosomes (haploidy), with < 46 chromosomes (hypodiploidy), or with 66 to 68 chromosomes (near triploidy)
Leukemic cell karyotype with t(v;11q23) MLL (KMT2A) rearranged, including t(4;11)/KMT2A-AF4
Leukemic cell karyotype t(5;14)/IL3-IG
Leukemic cell karyotype t(8;14), t(8;22), t(2;8) C-MYC rearranged
Presence of the Philadelphia (Ph) chromosome t(9;22) BCR-ABL1
Increased age in adults
BCR/ABL1-like molecular signature
Regardless of prognostic factors, the likelihood of initial remission is ≥ 95% in children and 70 to 90% in adults. Of children, > 80% have continuous disease-free survival for 5 years and appear to be cured. Of adults, < 50% have long-term survival. Factors contributing to poorer clinical outcomes in adults compared with children include the following:
Less ability to tolerate intensive chemotherapy
More frequent and severe comorbidities
Higher risk ALL genetics that confer chemotherapy resistance
Poorer adherence to ALL treatment regimens, which include frequent (often daily or weekly) out-patient chemotherapy and doctor visits
Less frequent use of pediatric-inspired treatment regimens
Most investigatory protocols select patients with poor prognostic factors for more intense therapy because the increased risk of and toxicity from treatment are outweighed by the greater risk of treatment failure leading to death.
Key Points
Acute lymphoblastic leukemia (ALL) is the most common cancer in children but also occurs in adults.
Central nervous system (CNS) involvement is common; most patients receive intrathecal chemotherapy and corticosteroids and sometimes CNS radiation therapy.
Response to treatment is good in children, with cure possible in > 80% of children but in < 50% of adults.
Repeat induction chemotherapy, immunotherapy, and stem cell transplantation may be helpful for relapse.
More Information
The following English-language resource may be useful. Please note that THE MANUAL is not responsible for the content of this resource.
Leukemia and Lymphoma Society: Resources for Healthcare Professionals: Provides information on education programs and conferences and resources for referrals to specialty care





