

Sturge-Weber syndrome is a congenital vascular disorder characterized by a facial port-wine nevus, a leptomeningeal capillary-venous malformation (leptomeningeal angioma), glaucoma, and neurologic complications (eg, seizures, focal neurologic deficits, intellectual disability). Diagnosis is clinical. Treatment is symptomatic.
Sturge-Weber syndrome is a neurocutaneous syndrome that occurs in 1 in 20,000 to 50,000 people (1). It is not inherited.
General reference
1. MedlinePlus [Internet]. Bethesda (MD): National Library of Medicine (US); [updated 2020 Jun 24]. Sturge-Weber syndrome; [updated 2018 Oct 1; reviewed 2018 Jun 01; cited 2025 Nov 11]; [about 5 p.]. Available from: Sturge-Weber syndrome: MedlinePlus Genetics.
Pathogenesis of Sturge-Weber Syndrome
Sturge-Weber syndrome is most commonly caused by a somatic mutation in the GNAQ gene on chromosome 9q21 and less commonly by somatic mutations in the related genes GNA11 and GNB2 (1–3). These mutations affect neural crest–derived endothelial cells during embryonic development. The GNAQ mutation causes constitutive G-protein activation, which leads to aberrant signaling through MAPK/ERK, P13K/AKT/mTOR, and Hippo-YAP pathways that results in impaired endothelial differentiation and progressive abnormal capillary-venous malformations in the brain, skin, and eyes. In the brain, these impairments manifest as leptomeningeal angiomatosis with impaired central venous drainage and progressive venous congestion, which lead to chronic hypoperfusion to the brain, which predisposes to seizures, stroke-like episodes, and progressive neurologic deterioration. The predilection for facial skin involvement, particularly in the distribution of the ophthalmic branch (V1) of the trigeminal nerve, and ipsilateral brain involvement shows the embryologic origin from neural crest cells emanating from the forebrain region.
A port-wine nevus (or sometimes a stain or birth mark) is a capillary malformation that typically develops on the forehead and upper eyelid in the distribution of the first and/or second division of the trigeminal nerve in patients with Sturge-Weber syndrome.
A similar vascular lesion, leptomeningeal capillary-venous malformation (previously called leptomeningeal angioma), can be present when a typical port-wine nevus involves the upper and lower eyelids on one side of the face. Ipsilateral angiomas occur in up to 30% of patients (4).
Usually, the nevus and leptomeningeal capillary-venous malformation are unilateral. Rarely, patients can have bilateral port-wine nevi in the distribution of the first division of the trigeminal nerve and bilateral leptomeningeal capillary-venous malformations.
A port-wine nevus may sometimes occur without a leptomeningeal capillary-venous malformation and its accompanying neurologic signs; in such cases, the eyes and eyelids may or may not be involved. Rarely, a leptomeningeal capillary-venous malformation occurs without the port-wine nevus and ocular involvement.
Neurologic complications of Sturge-Weber syndrome include seizures, focal neurologic deficits (eg, hemiparesis), and intellectual disability.
Sturge-Weber syndrome can also cause glaucoma and cerebral vascular narrowing, which may increase the risk of stroke due to thrombosis, venous occlusion, or infarction.
Pathogenesis references
1. MedlinePlus [Internet]. Bethesda (MD): National Library of Medicine (US); [updated 2020 Jun 24]. Sturge-Weber syndrome; [updated 2018 Oct 1; reviewed 2018 Jun 01; cited 2025 Nov 11]; [about 5 p.]. Available from: Sturge-Weber syndrome: MedlinePlus Genetics.
2. El Hachem M, Diociaiuti A, Galeotti A, et al. Multidisciplinary, multicenter consensus for the care of patients affected with Sturge-Weber syndrome. Orphanet J Rare Dis. 2025;20(1):28. Published 2025 Jan 16. doi:10.1186/s13023-024-03527-w
3. Fjær R, Marciniak K, Sundnes O, et al. A novel somatic mutation in GNB2 provides new insights to the pathogenesis of Sturge-Weber syndrome. Hum Mol Genet. 2021;30(21):1919-1931. doi:10.1093/hmg/ddab144
4. Bar C, Pedespan JM, Boccara O, et al. Early magnetic resonance imaging to detect presymptomatic leptomeningeal angioma in children with suspected Sturge-Weber syndrome. Dev Med Child Neurol. 2020;62(2):227-233. doi:10.1111/dmcn.14253
Symptoms and Signs of Sturge-Weber Syndrome
The port-wine nevus can vary in size and color, ranging from light pink to deep purple.

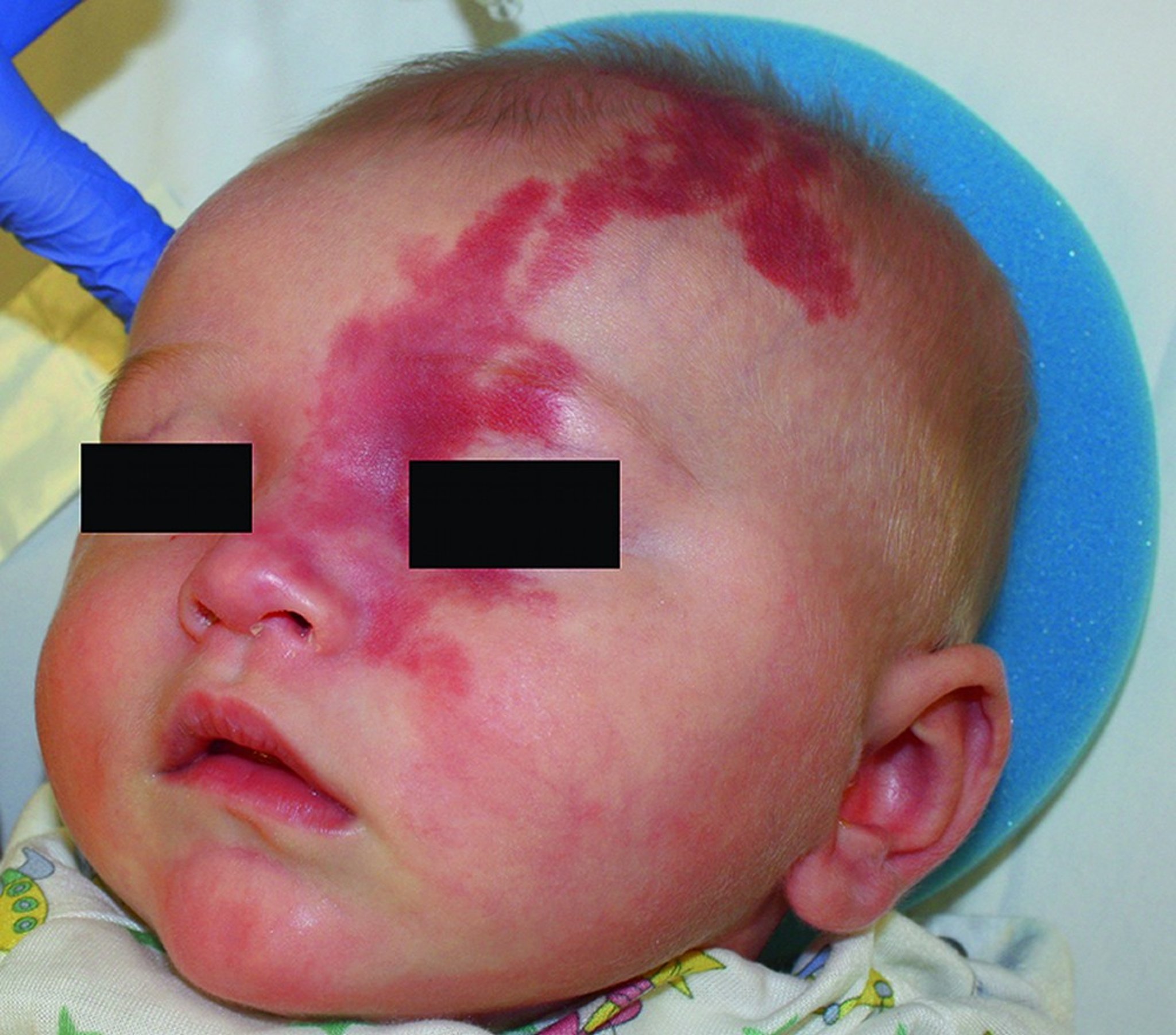
This photo shows an infant with a port-wine stain involving the ophthalmic branch (V1) of the trigeminal nerve.
© Springer Science+Business Media
Usually, the nevus and leptomeningeal capillary-venous malformation are unilateral. Rarely, patients can have bilateral port-wine nevi in the distribution of the first division of the trigeminal nerve and bilateral leptomeningeal capillary-venous malformations.
A port-wine nevus may sometimes occur without a leptomeningeal capillary-venous malformation and its accompanying neurologic signs; in such cases, the eyes and eyelids may or may not be involved. Rarely, a leptomeningeal capillary-venous malformation occurs without the port-wine nevus and ocular involvement. Epileptic seizures can occur in approximately 75 to 90% of patients (1); they typically start by age 1 year (2). Seizures are usually focal in nature but can become generalized. Hemiparesis may occur and can be transient or permanent (3). Hemiparesis of the side opposite the port-wine nevus can occur in up to approximately 60% of patients (4). Sometimes the hemiparesis worsens, especially in patients whose seizures cannot be controlled.
Approximately 50% of patients have intellectual disability, and more have some kind of learning difficulty (4). Development may be delayed.
Glaucoma may be present at birth or develop later. The eyeball may enlarge and bulge out of its socket (buphthalmos).
Vascular malformations may also occur in the oral mucosa. Dental manifestations (eg, gingival hyperplasia, intraoral angiomatosis) have been reported in up to 50% of patients with Sturge-Weber syndrome (5).
Symptoms and signs references
1. Disse SC, Küpper H, Bock A, et al. The natural history of pediatric Sturge-Weber Syndrome: A multinational cross-sectional study. Eur J Paediatr Neurol. 2025;54:200-209. doi:10.1016/j.ejpn.2025.02.004
2. Bar C, Kaminska A, Nabbout R. Spikes might precede seizures and predict epilepsy in children with Sturge-Weber syndrome: A pilot study. Epilepsy Res. 2018;143:75-78. doi:10.1016/j.eplepsyres.2018.03.020
3. Tillmann RP, Ray K, Aylett SE. Transient episodes of hemiparesis in Sturge Weber Syndrome - Causes, incidence and recovery. Eur J Paediatr Neurol. 2020;25:90-96. doi:10.1016/j.ejpn.2019.11.001
4. Powell S, Fosi T, Sloneem J, Hawkins C, Richardson H, Aylett S. Neurological presentations and cognitive outcome in Sturge-Weber syndrome. Eur J Paediatr Neurol. 2021;34:21-32. doi:10.1016/j.ejpn.2021.07.005
5. Pagin O, Del Neri NB, Battisti Mde P, Capelozza AL, Santos PS. Periodontal manifestations and ambulatorial management in a patient with Sturge-Weber syndrome. J Craniofac Surg. 2012;23(6):1809-1811. doi:10.1097/SCS.0b013e318271016c
Diagnosis of Sturge-Weber Syndrome
Brain MRI or head CT
The diagnosis of Sturge-Weber syndrome is suspected in infants or children with a characteristic port-wine nevus, especially when in the distribution of the ophthalmic branch of the trigeminal nerve. Neuroimaging helps confirm the diagnosis with the presence of leptomeningeal capillary-venous malformations.
If feasible, a comprehensive pediatric neurologic evaluation is recommended in the first 3 months of life for children with cutaneous manifestations of Sturge-Weber syndrome to check for neurologic complications (1). An EEG is also recommended to identify epilepsy. Routine follow-up evaluations with a neurologist are also recommended every 6 months for the first 2 years of life.
An ophthalmologic examination is done to check for eye complications (eg, glaucoma, retinal vascular malformations).
Dental evaluations should be performed as soon as the diagnosis is established.


This contrast-enhanced axial T1-weighted MR image shows typical cortical enhancement suggesting angiomatosis in Sturge-Weber syndrome.
Living Art Enterprises, LLC/Science Source/SCIENCE PHOTO LIBRARY
Brain MRI with contrast is used to check for a leptomeningeal capillary-venous malformation, but the malformation may not yet be appreciable on imaging in very young children. If MRI is not available, head CT may be done. CT may show calcifications in the cortex, but it is limited by low sensitivity (1).
Diagnosis reference
1. El Hachem M, Diociaiuti A, Galeotti A, et al. Multidisciplinary, multicenter consensus for the care of patients affected with Sturge-Weber syndrome. Orphanet J Rare Dis. 2025;20(1):28. Published 2025 Jan 16. doi:10.1186/s13023-024-03527-w
Treatment of Sturge-Weber Syndrome
Symptomatic treatment
The treatment of Sturge-Weber syndrome is focused on symptoms (1).
Antiseizure medications and medications to treat glaucoma are used. Sometimes hemispherectomy is done if patients have intractable seizures.
Low-dose aspirin is usually given, starting at the time of diagnosis, to help prevent seizures and strokes and possibly reduce progressive hemispheric atrophy by preventing sludging in the abnormal capillaries (Low-dose aspirin is usually given, starting at the time of diagnosis, to help prevent seizures and strokes and possibly reduce progressive hemispheric atrophy by preventing sludging in the abnormal capillaries (2, 3).
Selective photothermolysis (pulsed-dye laser) can lighten the port-wine nevus and is considered the standard of care in resource-rich settings (4).
Surgical procedures (ie, trabeculotomy, shunt placement) to alleviate intraocular pressure may be needed to control glaucoma effectively (5). Trabeculotomy is the most efficacious procedure.
Treatment references
1. Sabeti S, Ball KL, Bhattacharya SK, et al. Consensus Statement for the Management and Treatment of Sturge-Weber Syndrome: Neurology, Neuroimaging, and Ophthalmology Recommendations. Pediatr Neurol. 2021;121:59-66. doi:10.1016/j.pediatrneurol.2021.04.013
2. Lance EI, Sreenivasan AK, Zabel TA, Kossoff EH, Comi AM. Aspirin use in Sturge-Weber syndrome: side effects and clinical outcomes. J Child Neurol. 2013;28(2):213-218. doi:10.1177/0883073812463607
3. Bay MJ, Kossoff EH, Lehmann CU, Zabel TA, Comi AM. Survey of aspirin use in Sturge-Weber syndrome. J Child Neurol. 2011;26(6):692-702. doi:10.1177/0883073810388646
4. Sabeti S, Ball KL, Burkhart C, et al. Consensus Statement for the Management and Treatment of Port-Wine Birthmarks in Sturge-Weber Syndrome. JAMA Dermatol. 2021;157(1):98-104. doi:10.1001/jamadermatol.2020.4226
5. Karaconji T, Ting ER, Zagora SL, Ardern-Holmes S, Jamieson RV, Grigg JR. Surgical Treatment for SWS Glaucoma: Experience From a Tertiary Referral Pediatric Hospital. J Glaucoma. 2020;29(12):1132-1137. doi:10.1097/IJG.0000000000001645
Key Points
Sturge-Weber syndrome is a congenital vascular disorder characterized by a facial port-wine nevus, a leptomeningeal capillary-venous malformation, and neurologic complications.
Seizures occur in approximately 75 to 90% of patients and typically start by age 1 year.
Approximately 50% of patients have intellectual disability, and more have some kind of learning difficulty.
Diagnosis is by noting the characteristic port-wine nevus and performing brain imaging to check for a leptomeningeal capillary-venous malformation.
Treatment is often symptomatic with antiseizure medications and low-dose aspirin to help prevent strokes.Treatment is often symptomatic with antiseizure medications and low-dose aspirin to help prevent strokes.





