


La fibrosi cistica è una patologia ereditaria che provoca la produzione anomal di secrezioni dense da parte di alcune ghiandole, con conseguente danno a organi e tessuti, soprattutto nei polmoni e nel tratto digerente.
La fibrosi cistica è causata da varianti genetiche ereditarie che provocano secrezioni dense e appiccicose che ostruiscono i polmoni e altri organi.
I sintomi tipici includono gonfiore addominale, feci molli e scarso aumento di peso nei neonati, nonché tosse, respiro sibilante e frequenti infezioni del tratto respiratorio più avanti nel corso della vita.
La diagnosi si basa sui risultati del test del sudore e/o di test genetici.
Negli Stati Uniti, circa la metà dei soggetti con questa malattia è costituita da adulti.
I trattamenti consistono in antibiotici, broncodilatatori, farmaci per fluidificare le secrezioni polmonari e trattamenti per liberare le vie respiratorie per i problemi respiratori; integratori degli enzimi pancreatici e vitamine per i problemi digestivi, nonché farmaci per migliorare la funzione della proteina della fibrosi cistica nei soggetti che presentano certe varianti genetiche.
In alcuni soggetti è utile il trapianto di fegato e polmoni.
La fibrosi cistica è la condizione ereditaria più frequente che riduce la durata di vita tra i soggetti di razza bianca. Negli Stati Uniti, si manifesta in circa in 1 su 3.300 neonati di razza bianca e in 1 su 15.300 di quelli di razza nera. Non è frequente negli asiatici. Poiché i miglioramenti terapeutici hanno prolungato l’aspettativa di vita dei malati di fibrosi cistica, circa la metà di questi malati negli Stati Uniti è costituita da adulti. La fibrosi cistica colpisce indistintamente entrambi i sessi.
Fibrosi cistica: non solo una malattia polmonare
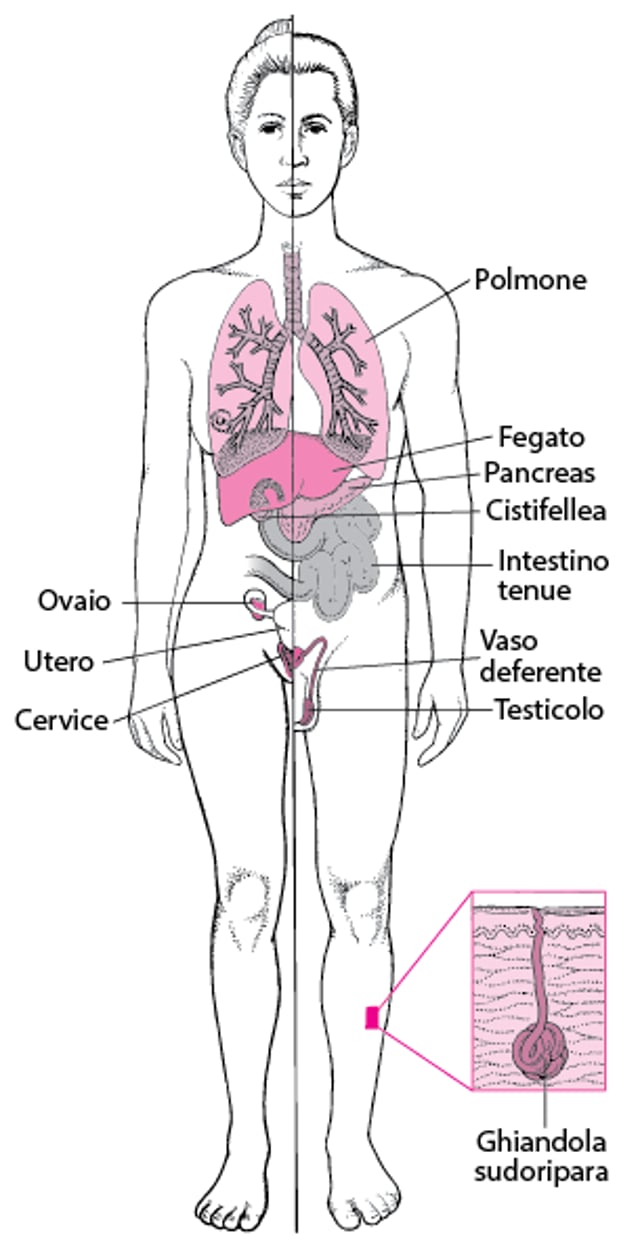
La fibrosi cistica colpisce i polmoni e vari altri organi. Nei polmoni, dense secrezioni bronchiali ostruiscono le piccole vie aeree, che diventano infette e si infiammano. Quando la malattia evolve, le pareti bronchiali si ispessiscono, le vie aeree si riempiono di secrezioni infette, le aree del polmone si contraggono e si verifica un ingrossamento linfonodale. Nel fegato, le secrezioni dense ostruiscono i dotti biliari. Possono svilupparsi calcoli della cistifellea. Nel pancreas, le secrezioni dense possono ostruire completamente la ghiandola impedendo agli enzimi digestivi di raggiungere l’intestino. Il pancreas può produrre meno insulina, causando la comparsa del diabete in alcuni soggetti (generalmente nell’adolescenza o nell’età adulta). Nell’intestino tenue, l’ileo da meconio (un tipo di occlusione intestinale) può essere dovuta a secrezioni dense e richiede un intervento chirurgico in alcuni neonati. Gli organi riproduttivi sono interessati dalla fibrosi cistica in vari modi, causando spesso sterilità maschile. Le ghiandole sudoripare producono un sudore più salato del normale. |

Cause della fibrosi cistica
Geni anomali
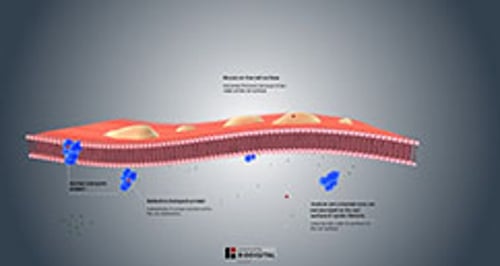
La fibrosi cistica insorge quando un soggetto eredita due copie difettose (varianti) di un particolare gene, una da ciascun genitore. Questo gene è chiamato regolatore della conduttanza transmembrana della fibrosi cistica (cystic fibrosis transmembrane conductance regulator, CFTR). Esistono diverse varianti del gene CFTR. Per esempio, quella più comune è chiamata variante F508del. Il gene CFTR controlla la produzione di una proteina che regola il movimento di cloruro, bicarbonato e sodio (sale) attraverso le membrane cellulari. Le varianti del gene CFTR causano disfunzioni della proteina. Se la proteina non funziona correttamente, il movimento di cloruro, bicarbonato e sodio subisce alterazioni che causano l’ispessimento e un aumento dell’appiccicosità delle secrezioni in tutto il corpo.
Nel mondo, circa 3 soggetti di razza bianca su 100 sono portatori di una copia difettosa del gene CFTR. I soggetti con una copia difettosa sono portatori ma non hanno la malattia. Circa 3 soggetti di razza bianca su 10.000 ereditano due copie del gene difettoso e sviluppano la fibrosi cistica.
Secrezioni anomale
La fibrosi cistica interessa molti organi in tutto l’organismo e quasi tutte le ghiandole che secernono liquidi all’interno di un dotto (ghiandole esocrine).
Gli organi interessati più spesso sono:
polmoni
Pancreas
Intestino
Fegato e cistifellea
Organi riproduttivi
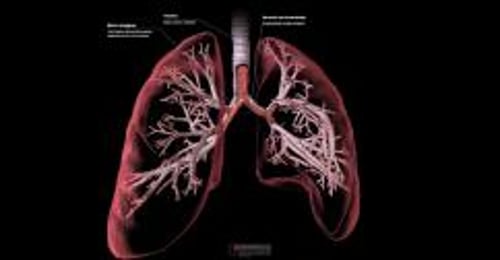
I polmoni sono normali alla nascita, ma successivamente possono presentarsi problemi in qualunque momento quando le secrezioni dense iniziano a ostruire le piccole vie aeree (cosiddetti tappi di muco). La formazione dei tappi causa infezioni batteriche croniche e infiammazioni che danneggiano permanentemente le vie aeree (cosiddetta bronchiectasia). Questi problemi rendono la respirazione sempre più difficile e riducono la capacità del polmone di trasferire l’ossigeno al sangue. Si sviluppano anche frequenti infezioni batteriche del tratto respiratorio che interessano i seni paranasali.
L’ostruzione dei dotti del pancreas impedisce agli enzimi digestivi di raggiungere l’intestino. L’assenza di questi enzimi causa scarso assorbimento dei grassi, delle proteine e delle vitamine (malassorbimento). Questo scarso assorbimento a sua volta può causare deficit nutrizionali e scarsa crescita. Alla fine, possono formarsi delle cicatrici nel pancreas, che non produce più abbastanza insulina, causando in alcune persone lo sviluppo del diabete. Tuttavia, il 5-15% circa dei pazienti affetti da fibrosi cistica e portatori di certe varianti non sviluppa problemi digestivi a livello del pancreas.
L’intestino può essere ostruito dalle secrezioni dense. Questo blocco è comune subito dopo la nascita, perché il contenuto del tratto digerente del feto (chiamato meconio) è insolitamente denso. Tale ostruzione dell’intestino tenue è definita ileo da meconio e nel colon è definita sindrome da tappo di meconio. I bambini più grandi e gli adulti possono anche avere problemi di stipsi e ostruzione intestinale (detta sindrome da ostruzione intestinale distale).
Fegato e cistifellea possono essere bloccati dalle secrezioni dense, che alla fine possono provocare cicatrizzazione del fegato (fibrosi). Possono formarsi calcoli biliari.
Le secrezioni dense possono bloccare gli organi riproduttivi con conseguente infertilità, soprattutto negli uomini, e molto meno nelle donne.
Le ghiandole sudoripare secernono fluidi contenenti più sale del normale, aumentando il rischio di disidratazione.
Sintomi della fibrosi cistica
I sintomi della fibrosi cistica possono variare a seconda dell’età della persona.
Neonati e bambini in tenera età
Il 10% circa dei neonati con fibrosi cistica presenta ileo da meconio, che causa vomito, gonfiore (distensione) addominale e assenza di movimenti intestinali. L’ileo da meconio talvolta è complicato dalla perforazione intestinale, una grave condizione che causa infezione e peritonite (infiammazione del tessuto che riveste la cavità e gli organi addominali) e, se non trattata, shock e decesso. Alcuni neonati presentano torsione dell’intestino su sé stesso (volvolo) o sviluppo incompleto dell’intestino. I neonati con ileo da meconio quasi sempre sviluppano successivamente altri sintomi della fibrosi cistica.
Spesso il primo sintomo di fibrosi cistica in un neonato che non presenta un ileo da meconio è un ritardo della capacità di riacquistare peso alla nascita, oppure un insufficiente incremento di peso a 4-6 settimane di vita. Questo scarso aumento di peso è dovuto a scarso assorbimento delle sostanze nutritive legato a quantità inadeguate di enzimi pancreatici. Spesso il neonato presenta feci frequenti, abbondanti, maleodoranti e oleose, talvolta con gonfiore (distensione) addominale. Senza trattamento, l’aumento di peso nei neonati e nei bambini più grandi è lento a dispetto di un appetito normale o forte.
Bambini più grandi e adulti
Se la diagnosi non viene posta durante lo screening neonatale, la metà circa dei bambini con fibrosi cistica viene condotta inizialmente dal medico per la presenza di tosse frequente, respiro sibilante e infezioni dell’apparato respiratorio. La tosse, che rappresenta il sintomo più rilevante, spesso è accompagnata da vomito e alterazioni del sonno. I bambini hanno difficoltà a respirare, respiro sibilante o entrambi. Quando la malattia evolve, i bambini sviluppano una tolleranza sempre minore per l’esercizio fisico, le infezioni polmonari tendono a comparire più frequentemente, il torace diventa a forma di botte e l’insufficienza di ossigeno può determinare l’ispessimento delle dita ( see page Ippocratismo) e un colore bluastro dei letti ungueali. Si possono formare polipi nasali. I seni paranasali possono riempirsi di secrezioni dense che causano sinusiti croniche o ricorrenti.
I bambini più grandi e gli adulti possono avere episodi di stipsi o sviluppare ostruzione ricorrente e a volte cronica dell’intestino. I sintomi includono un cambiamento delle caratteristiche delle feci, crampi addominali, scarso appetito e a volte vomito. Il reflusso gastroesofageo è abbastanza comune tra i bambini più grandi e negli adulti.
Quando un bambino o un adulto affetto da fibrosi cistica suda eccessivamente per l’alta temperatura esterna o a causa della febbre, può disidratarsi in seguito alla perdita di acqua e sale. Un genitore può osservare la formazione di cristalli di sale o perfino un sapore salato sulla cute del bambino.
Gli adolescenti spesso presentano un rallentamento della crescita e pubertà ritardata. Con l’evoluzione della malattia, il problema fondamentale è rappresentato dalle infezioni polmonari. Le infezioni polmonari ricorrenti distruggono gradualmente i polmoni.
Complicanze della fibrosi cistica
La fibrosi cistica presenta molte complicanze.
L’assorbimento insufficiente delle vitamine liposolubili A, D, E e K determina talvolta cecità notturna, osteopenia (riduzione della densità ossea), osteoporosi, anemia e disturbi emorragici. In lattanti e bambini molto piccoli non trattati si può osservare la fuoriuscita parziale del retto attraverso l’orifizio anale, una condizione definita prolasso rettale. I neonati con fibrosi cistica alimentati con latte ipoallergenico o con latte di soia possono sviluppare anemia e gonfiore agli arti, per un malassorbimento proteico.
Le complicanze della fibrosi cistica negli adolescenti e negli adulti comprendono la rottura delle piccole cavità polmonari (alveoli) nella cavità pleurica (lo spazio tra il polmone e la parete toracica), consentendo la penetrazione dell’aria in questo spazio (pneumotorace) e causando il collasso del polmone. Altre complicanze comprendono insufficienza cardiaca e sanguinamento massivo o ricorrente nelle vie respiratorie.
Circa il 2% dei bambini, il 20% degli adolescenti e fino al 50% degli adulti con fibrosi cistica sviluppano diabeteinsulino-dipendente, perché il pancreas danneggiato non produce più una quantità sufficiente di insulina.
L’ostruzione dei dotti biliari dovuta alle secrezioni dense può determinare infiammazione e infine cicatrizzazione del fegato (cirrosi) nel 3-4% circa degli adulti con fibrosi cistica. La cirrosi può aumentare la pressione nelle vene che entrano nel fegato (ipertensione portale), causando una dilatazione e un indebolimento delle vene nella parte inferiore dell’esofago (varici esofagee), che possono rompersi e sanguinare abbondantemente.
In quasi tutti i soggetti con fibrosi cistica la cistifellea è piccola, piena di bile densa e malfunzionante. Circa il 10% dei soggetti sviluppa calcoli biliari, ma solo una piccola percentuale è sintomatica. La rimozione chirurgica della cistifellea è raramente necessaria.
I soggetti con fibrosi cistica spesso hanno alterazioni della funzione riproduttiva. Quasi tutti gli uomini presentano una conta spermatica bassa o assente (che li rende sterili), perché uno dei dotti testicolari (dotto deferente) si è sviluppato in modo anomalo, bloccando il passaggio dello sperma. Nelle donne, le secrezioni della cervice uterina sono troppo dense e riducono la fertilità. Tuttavia, molte donne con fibrosi cistica hanno portato a termine la gravidanza. L’esito della gravidanza per madre e neonato dipende dallo stato di salute della madre durante la gravidanza. D’altro canto, la funzione sessuale maschile o femminile non è interessata.
Altre complicanze possono includere artrite, dolore cronico, problemi del sonno e apnea ostruttiva nel sonno, calcoli renali, malattia renale, depressione e ansia, perdita uditiva neurosensoriale e ronzii nell'orecchio (tinnito) causati dall'esposizione a farmaci che danneggiano le orecchie (specialmente aminoglicosidi) e un aumento del rischio di tumore dei dotti biliari, del pancreas e dell’intestino.
Diagnosi di fibrosi cistica
Test di screening neonatale
Esame del sudore
Test genetici
Screening dei portatori sani
Altri esami
Screening neonatale
Negli Stati Uniti, tutti i neonati sono sottoposti a test di screening per la fibrosi cistica. Si misura il livello di tripsina (un enzima pancreatico che interviene nella digestione ) in una goccia di sangue prelevata su carta da filtro. Se il livello di tripsina nel sangue è alto, i neonati sono sottoposti a test di conferma, che include l’analisi del sudore e/o test genetici. Molti casi di fibrosi cistica vengono ora identificati grazie ai test di screening neonatale.
Se lo screening neonatale non viene eseguito, la diagnosi di fibrosi cistica è di solito confermata durante l’infanzia o la prima fanciullezza, ma nel 10% circa dei casi la causa rimane sconosciuta fino all’adolescenza o alla prima età adulta.
Esame del sudore
Un esame del sudore viene eseguito sui neonati con test di screening positivo e nei lattanti, bambini e soggetti più grandi con sintomi che suggeriscono la fibrosi cistica. Questo test, eseguito in ambulatorio, misura la quantità di sale nel sudore. La pilocarpina viene applicata sulla cute per stimolare la sudorazione; il sudore viene quindi raccolto da una carta bibula o con un tubicino sottile premuto sulla cute. Si misura quindi la concentrazione di sale nel sudore. Una concentrazione di sale superiore ai valori normali conferma la diagnosi nei soggetti con sintomi di fibrosi cistica o con fratelli affetti da tale patologia. Sebbene i risultati del test siano validi in un neonato con più di 48 ore di vita, risulta talvolta difficile raccogliere una quantità di sudore sufficiente in neonati con meno di 2 settimane di vita.
Test genetici
Nei neonati positivi al test di screening neonatale, in soggetti che presentano uno o più sintomi caratteristici o persone con familiarità con la patologia, la diagnosi di fibrosi cistica può essere facilitata anche da test genetici per anomalie del gene CFTR. La presenza di due geni della fibrosi cistica alterati (varianti) è compatibile con la diagnosi di fibrosi cistica. Tuttavia, per confermare la diagnosi è comunque necessario un test del sudore positivo. Inoltre, poiché i test genetici tipici non valutano tutte le oltre 2.000 diverse varianti della fibrosi cistica, il fatto di non individuare due varianti non garantisce che il soggetto non sia affetto da fibrosi cistica (sebbene la probabilità di avere la malattia sia molto bassa). La malattia può essere diagnosticata prima della nascita eseguendo un test genetico sul feto con un prelievo dei villi coriali o con l’amniocentesi.
Alcuni bambini con test di screening neonatale positivo per la fibrosi cistica possono essere difficili da classificare, anche dopo il test del sudore o i test genetici. Questi bambini non hanno sintomi correlati alla fibrosi cistica, con risultati del test del sudore al limite della positività e della negatività e non presentano varianti genetiche della fibrosi cistica o ne presentano solo una. I medici diagnosticano questo gruppo come affetto da sindrome metabolica correlata a CFTR (CFTR-related metabolic syndrome, CRMS), chiamata anche diagnosi inconcludente di fibrosi cistica positiva allo screening (cystic fibrosis screen positive inconclusidve diagnosis, CFSPID). Sebbene la maggior parte di questi neonati rimanga sana, nel corso della vita il 10% circa sviluppa sintomi correlati alla fibrosi cistica e viene loro diagnosticata la malattia o un disturbo correlato. Pertanto, tutti questi bambini devono essere monitorati regolarmente in un centro di cura per la fibrosi cistica.
Alcuni soggetti, di solito adulti, sviluppano sintomi che interessano solo un organo, spesso con un risultato del test del sudore intermedio e senza le due varianti che causano la malattia. Per esempio, i sintomi possono interessare solo il pancreas (provocando pancreatite), i polmoni (provocando bronchiectasia) o gli organi riproduttivi maschili (causando infertilità). Vengono diagnosticati come disturbo correlato a CFTR.
Test del portatore
Il test del portatore può essere svolto sui soggetti che vogliono diventare genitori o si sottopongono ad assistenza prenatale. In particolare, i familiari di un bambino affetto da fibrosi cistica talvolta desiderano informarsi sulle probabilità di avere figli con tale patologia, per cui è necessario offrire il test e la consulenza genetici. Si preleva un campione di sangue per cercare di scoprire se il soggetto possiede un gene della fibrosi cistica difettoso (variante).
Salvo il caso in cui entrambi i potenziali genitori abbiano almeno una di queste varianti, i figli non saranno colpiti da tale patologia. Se entrambi i genitori sono portatori di un gene della fibrosi cistica difettoso, ciascuna gravidanza ha una possibilità del 25% di far nascere un bambino affetto da fibrosi cistica, del 50% di far nascere un bambino portatore e del 25% di far nascere un bambino non portatore.
Altri esami
Poiché la fibrosi cistica può interessare diversi organi, può essere utile eseguire altri test. I livelli degli enzimi pancreatici di solito risultano ridotti e l’analisi delle feci può evidenziare livelli bassi o non rilevabili di elastasi (enzima digestivo secreto dal pancreas) e un elevato livello di grassi.
Gli esami del sangue servono a determinare l’eventuale riduzione della secrezione di insulina e l’aumento della glicemia. Si effettuano anche esami del sangue per ricercare problemi a carico del fegato e per misurare i livelli di vitamine liposolubili.
I medici eseguono regolarmente prelievi di campioni di materiale dalla gola o di espettorato dei polmoni e li mettono in coltura per identificare i batteri presenti nelle vie aeree e decidere quali antibiotici possono essere necessari.
I test di funzionalità polmonare possono evidenziare una compromissione della respirazione e sono validi indicatori della funzionalità polmonare. Questi esami vengono eseguiti diverse volte all’anno e ogniqualvolta vi sia un declino della salute del soggetto.
Inoltre, la radiografia toracica e la tomografia computerizzata (TC) del torace possono rivelarsi utili nel documentare l’infezione e l’entità del danno polmonare. La TC delle cavità nasali viene eseguita nei soggetti con gravi sintomi sinusali, specie se presentano polipi nasali o se si deve valutare il trattamento chirurgico.
Prognosi della fibrosi cistica
La gravità della fibrosi cistica varia enormemente secondo il soggetto, indipendentemente dall’età. La gravità è determinata ampiamente dalla misura in cui sono interessati i polmoni. Negli Stati Uniti si stima che i soggetti con fibrosi cistica nati nel 2019 abbiano un’aspettativa di vita fino a circa 48 anni di età. La prospettiva di una sopravvivenza più lunga è migliorata costantemente negli ultimi 50 anni, soprattutto grazie alla diagnosi più precoce e alle terapie attuali in grado di posticipare alcuni dei cambiamenti che si verificano a livello polmonare. La sopravvivenza a lungo termine è significativamente migliore in soggetti che non sviluppano problemi al pancreas.
Ciononostante, il peggioramento è inevitabile e conduce alla perdita della funzione polmonare e, alla fine, alla morte. Nei soggetti con fibrosi cistica spesso la morte sopraggiunge per insufficienza respiratoria dopo molti anni di peggioramento della funzione polmonare. Tuttavia, in un numero esiguo di casi, essa è dovuta a insufficienza cardiaca, patologia epatica, emorragia nelle vie respiratorie o complicanze chirurgiche che includono il trapianto di polmoni e/o fegato. Nonostante i numerosi problemi, le persone affette da fibrosi cistica spesso frequentano la scuola o lavorano fino a poco tempo prima di morire.
Trattamento della fibrosi cistica
Vaccinazioni di routine
Antibiotici, farmaci per inalazione per diluire le secrezioni delle vie respiratorie e tecniche di disostruzione delle vie aeree per rimuovere le secrezioni
Farmaci che aiutano a prevenire il restringimento delle vie respiratorie (broncodilatatori) e talvolta corticosteroidi
Integratori di enzimi pancreatici e vitamine
Dieta ipercalorica
Nei soggetti con varianti specifiche, modulatori del CFTR
Un soggetto con fibrosi cistica deve seguire un programma terapeutico completo sotto la guida di uno specialista nella cura della patologia, di solito un pediatra o un internista, affiancato da un’équipe di altri medici e infermieri, un dietologo, un terapista respiratorio o fisioterapista e idealmente un assistente sociale, un consulente genetico, un farmacista e un operatore di salute mentale. Gli obiettivi della terapia comprendono prevenzione a lungo termine e trattamento di problemi polmonari e digestivi e di altre complicanze, mantenimento di una buona nutrizione e attività fisica.
I bambini con fibrosi cistica necessitano di supporto socio-psicologico perché talvolta non sono in grado di partecipare alle normali attività infantili e rischiano di sentirsi isolati. Le maggiori responsabilità del trattamento di un bambino affetto da fibrosi cistica gravano sui genitori, che devono essere adeguatamente informati e preparati per comprendere la malattia e le motivazioni alla base del trattamento.
Gli adolescenti necessitano di guida ed educazione nel passaggio all’indipendenza e all’assunzione delle responsabilità per le proprie cure.
Gli adulti necessitano di sostegno nell’affrontare i problemi correlati a occupazione, relazioni, assicurazione sanitaria e deterioramento della salute.
Trattamenti per i polmoni
Il trattamento dei problemi polmonari si concentra su
prevenzione del blocco delle vie aeree
controllo delle infezioni
Il paziente deve sottoporsi a tutte le vaccinazioni di routine, in special modo quelle per le infezioni che causano problemi respiratori, come Haemophilus influenzae, influenza, morbillo, pertosse, pneumococco e varicella. La vaccinazione anti-COVID-19 dovrebbe essere somministrata in base alle attuali raccomandazioni dei Centers for Disease Control and Prevention (CDC).
Le tecniche di disostruzione delle vie aeree, che includono drenaggio posturale, percussione del torace, vibrazione a mano sulla parete toracica e stimolazione della tosse ( see page Fisioterapia toracica) vanno avviate al momento della diagnosi di fibrosi cistica. I genitori di un bambino piccolo possono imparare queste tecniche e utilizzarle a casa quotidianamente. I bambini più grandi e gli adulti possono eseguire le tecniche di disostruzione autonomamente, usando appositi dispositivi per la respirazione, un dispositivo gonfiabile da indossare che vibra ad alta frequenza (detto dispositivo a oscillazione ad alta frequenza) o speciali manovre di respirazione. Gli esercizi aerobici regolari possono essere utili a mantenere libere le vie aeree.
I broncodilatatori sono farmaci che aiutano a prevenire il restringimento delle vie aeree. Di solito vengono assunti per inalazione. I soggetti con gravi forme polmonari e ridotta ossigenazione nel sangue possono necessitare di un’ossigenoterapia supplementare. In generale, i soggetti con insufficienza respiratoria cronica non traggono vantaggio dall’uso di un ventilatore (macchina respiratoria). Tuttavia, brevi periodi di ventilazione meccanica in ospedale possono aiutare in caso di infezione acuta, dopo un intervento chirurgico o in attesa di un trapianto di polmone.
I farmaci che aiutano a fluidificare il muco denso nelle vie aeree, come dornase alfa o soluzione fisiologica ipertonica (una soluzione salina altamente concentrata), sono ampiamente utilizzati. Questi farmaci vengono inalati attraverso un nebulizzatore. Facilitano l’espettorato, migliorano la funzione polmonare e possono anche ridurre la frequenza di infezioni gravi delle vie respiratorie.
I corticosteroidi, come prednisone o dexametasone, somministrati per via orale, possono alleviare i sintomi nei neonati con grave infiammazione bronchiale, in soggetti con vie aeree ristrette che non si riesce ad aprire con i broncodilatatori e nei soggetti con reazione polmonare allergica a un tipo di fungo (aspergillosi broncopolmonare allergica). Anche l’aspergillosi broncopolmonare allergica viene trattata con farmaci antimicotici somministrati per via orale, per via endovenosa o entrambe.
Talvolta, viene utilizzato l’ibuprofene, un farmaco antinfiammatorio non steroideo (FANS) per rallentare il deterioramento della funzione polmonare.
Sono necessari farmaci per trattare l’infiammazione cronica dei seni paranasali (sinusite), dato che questo problema è molto comune. Le opzioni terapeutiche includono lavaggio del naso con una soluzione salina (irrigazione nasale con soluzione salina), l'inalazione di dornasi alfa mediante un nebulizzatore e l’irrigazione di naso e seni paranasali con antibiotici. Per trattare l'infiammazione e il gonfiore delle mucose nasali (rinite allergica) è raccomandato uno spray nasale a base di corticosteroidi.
Antibiotici
Le infezioni respiratorie devono essere trattate il prima possibile con gli antibiotici. Al primo segno di infezione respiratoria, si raccoglie e si esamina un campione di espettorato o un tampone dal fondo della gola e dalle tonsille per identificare l’organismo infettante e perché il medico possa scegliere i farmaci più adatti per controllarla. Lo Staphylococcus aureus, inclusi i ceppi resistenti alla meticillina o sensibili alla meticillina, e le specie di Pseudomonas sono i patogeni riscontrati con maggiore frequenza. Per trattare le infezioni da stafilococco si possono somministrare molti antibiotici orali diversi. Per trattare un’infezione da Pseudomonas, si somministra una formulazione inalatoria di tobramicina, aztreonam o colistina per 4 settimane.
Tuttavia, se l’infezione è grave, può essere necessaria la somministrazione endovenosa, In questo trattamento prevede la combinazione di aminoglicoside tobramicina (o talvolta amikacina) con un altro antibiotico mirato in modo specifico allo Pseudomonas. Gli altri antibiotici includono cefalosporine, penicilline, fluorochinoloni e monobattami. Tale trattamento spesso richiede il ricovero, ma può essere effettuato a domicilio.
L’assunzione della forma inalabile della tobramicina o di aztreonam a mesi alterni per un periodo prolungato, nonché l’assunzione continua di una forma orale di azitromicina 3 volte alla settimana possono aiutare a controllare l’infezione da Pseudomonas e rallentare il deterioramento della funzione polmonare.
Modulatori di CFTR
I modulatori di CFTR sono farmaci orali assunti in maniera cronica che migliorano la funzione della proteina difettosa creata dalle varianti del gene CFTR.
Esistono quattro modulatori di CFTR o loro combinazioni per i soggetti che presentano specifiche varianti: ivacaftor, lumacaftor/ivacaftor, tezacaftor/ivacaftor e elexacaftor/tezacaftor/ivacaftor. Questi farmaci possono essere usati per trattare il 90% circa dei soggetti affetti da fibrosi cistica. I medici prescrivono tali farmaci ai soggetti in base all’età e alle varianti.
Ivacaftor è somministrato a pazienti di almeno 4 mesi di età che hanno almeno 1 copia di una variante specifica della fibrosi cistica.
La combinazione lumacaftor e ivacaftor può essere somministrata ai pazienti di almeno 2 anni di età che presentano 2 copie della variante F508del.
La combinazione tezacaftor e ivacaftor può essere somministrata ai pazienti di almeno 6 anni di età che presentano 2 copie della variante F508del o altre varianti specifiche.
La combinazione elexacaftor, tezacaftor e ivacaftor può essere somministrata ai pazienti di almeno 6 anni di età che presentano almeno 1 copia della variante F508del o 1 copia di alcune varianti rare.
I modulatori di CFTR possono migliorare la funzione polmonare, la funzione del pancreas e la qualità della vita; aumentare il peso, ridurre la concentrazione di sale nel sudore e la frequenza delle infezioni polmonari e dei ricoveri. Sebbene tutti questi farmaci possano essere utili, solo ivacaftor e la combinazione di elexacaftor, tezacaftor e ivacaftor è considerata una terapia altamente efficace.
Sono in corso studi per lo sviluppo di farmaci che aiutino i soggetti interessati da altre varianti che causano la fibrosi cistica.
Clisteri e ammorbidenti delle feci
I neonati con ostruzioni intestinali possono essere trattati con speciali soluzioni di clisteri, ma spesso richiedono trattamento chirurgico.
Bambini più grandi e adulti che soffrono di stipsi o blocco intestinale parziale possono essere trattati con ammorbidenti delle feci, clisteri e soluzioni speciali somministrate oralmente o attraverso un tubicino di plastica flessibile (sondino nasogastrico) passato nello stomaco attraverso il naso o la bocca.
Dieta e integratori alimentari
La dieta deve fornire una quota proporzionale di proteine e calorie sufficienti per una crescita normale. Poiché digestione e assorbimento possono essere anomali anche quando si usano integratori degli enzimi pancreatici, la maggior parte dei bambini ha bisogno di consumare il 30-50% di calorie in più rispetto alla quantità generalmente raccomandata per garantire una crescita adeguata. La quota di grassi deve essere normale o elevata. Gli integratori orali a elevato contenuto calorico possono fornire calorie aggiuntive per bambini e adulti.
I soggetti che non riescono ad assorbire dal cibo sostanze nutritive in quantità adeguata necessitano di un’integrazione alimentare con un sondino introdotto nello stomaco o nell’intestino tenue.
I soggetti con fibrosi cistica devono assumere una dose doppia rispetto a quella consigliata quotidianamente di vitamine liposolubili (A, D, E e K) in una formulazione speciale assorbita più facilmente.
I farmaci che stimolano l’appetito possono essere utili. In caso di attività fisica, febbre o esposizione ad alte temperature, i soggetti con fibrosi cistica devo aumentare l’apporto di sale e liquidi.
Integratori degli enzimi pancreatici
I soggetti con pancreas interessato dalla fibrosi cistica devono assumere capsule di integratori dell’enzima pancreatico con tutti i pasti e gli spuntini. Per i neonati, i genitori aprono le capsule e mescolano il contenuto con un cibo acido come la salsa di mele, in modo che lo speciale rivestimento dell’integratore degli enzimi pancreatici non si dissolva prima di raggiungere l’intestino. Per alcuni pazienti, farmaci che riducono l’acido gastrico, come un bloccante dell’istamina-2 o un inibitore della pompa protonica, possono migliorare l’efficacia degli enzimi pancreatici. Tipi di latte formulati in modo speciale, con grassi e proteine più digeribili, possono essere utili nei neonati con insufficienza pancreatica o crescita insufficiente.
Insulina
I soggetti affetti da fibrosi cistica e diabete devono ricorrere alle iniezioni di insulina. I farmaci orali per il diabete non rappresentano un trattamento adeguato.
Oltre all’insulina, nel controllo rientrano la consulenza nutrizionale, un programma di gestione autonoma del diabete e il monitoraggio per le complicanze a livello renale e oculare. Per tali soggetti è inoltre necessaria una speciale consulenza nutrizionale, poiché le raccomandazioni alimentari ordinarie valide per chi è affetto dal solo diabete o dalla sola fibrosi cistica non sono adeguate.
Intervento chirurgico
Talvolta, può rendersi necessario l’intervento chirurgico in caso di collasso polmonare, sinusite cronica, infezione cronica grave limitata a una singola area polmonare, sanguinamento dai vasi dell’esofago, malattie della cistifellea o occlusioni intestinali. È possibile trattare il sanguinamento polmonare massiccio e ricorrente con una metodica, definita embolizzazione, che blocca le arterie sanguinanti.
Il trapianto di fegato si è dimostrato efficace nei soggetti con emorragie causata da varici esofagee o grave danno epatico.
Il trapianto di entrambi i polmoni, in caso di malattie polmonari gravi, sta diventando più frequente e di maggiore successo, grazie all’esperienza e al miglioramento delle tecniche. Per gli adulti con fibrosi cistica la sopravvivenza media dopo un doppio trapianto polmonare è di circa 9 anni.
Altri trattamenti
I soggetti affetti da insufficienza cardiaca assumono farmaci (diuretici) per ridurre la ritenzione idrica. I diuretici aiutano aumentando la quantità di acqua eliminata dall’organismo attraverso i reni. I pazienti devono anche limitare l’assunzione di sale da tavola e cibi salati.
Le iniezioni di ormone della crescita umano possono migliorare la funzione polmonare, aumentare altezza e peso e ridurre il tasso di ricoveri. Tuttavia, questo farmaco è costoso e reca disturbi, quindi i medici in genere non lo prescrivono.
Alcune persone con bassi livelli di ossigeno nel sangue possono necessitare di ossigeno supplementare somministrato di solito attraverso una sonda (cannula) nasale a due vie con una maschera aderente posta sul naso o su naso e bocca. Attraverso la maschera si eroga una miscela di ossigeno e aria sotto pressione. Questa tecnica, chiamata pressione positiva delle vie aeree a doppio livello (BiPAP) o pressione continua positiva delle vie aeree (CPAP), può aiutare i pazienti a mantenere livelli di ossigeno normali mentre dormono.
Problematiche della fase terminale
I soggetti con fibrosi cistica e i loro familiari devono discutere con il proprio medico e con l’équipe sanitaria la prognosi e il tipo di trattamento che desiderano assumere. Queste discussioni sono particolarmente importanti per le persone la cui funzione polmonare sta peggiorando. I pazienti devono essere preparati a quello che li aspetta e sapere quali trattamenti possono essere somministrati per prolungare la vita. Con la fibrosi cistica in stadio avanzato pazienti e relative famiglie devono discutere i potenziali benefici e gli oneri del trapianto di polmone.
I medici devono fornire ai soggetti con fibrosi cistica le informazioni necessarie per prendere le decisioni sulle proprie cure e aiutarli a stabilire come e quando accettare la morte e come parlarne. La maggior parte dei malati di fibrosi cistica che devono affrontare la fine della vita si trova in un’età compresa fra la tarda adolescenza e età adulta, ed è responsabile e in grado di prendere decisioni.
Quando i trattamenti aggressivi non sono più efficaci, i medici possono somministrare trattamenti mirati solo ad alleviare i sintomi (cosiddette cure palliative), Generalmente è opportuno che il paziente esponga in largo anticipo la propria volontà relativa all’assistenza in fase terminale. Questo tipo di accordi anticipati sono molto importanti, perché, successivamente, la malattia spesso impedisce al soggetto di illustrare i propri desideri. Questo processo decisionale anticipato per l’assistenza in fase terminale viene detto pianificazione di assistenza anticipata e deve prevedere l’attuazione di opportuni documenti legali che riflettano i desideri della persona in relazione alle cure di fine vita.
Ulteriori informazioni
Di seguito si riportano alcune risorse in lingua inglese che possono essere utili. Si prega di notare che I MANUALI non sono responsabili del contenuto di tali risorse.
Cystic Fibrosis Foundation: una risorsa che offre informazioni sulle opzioni terapeutiche disponibili, lo sviluppo di farmaci, la ricerca e i servizi di supporto alla comunità
Centers for Disease Control and Prevention (CDC): Current recommendations for COVID-19 vaccination