




Amyloidosen sind eine Gruppe unterschiedlicher Krankheiten, denen die extrazelluläre Ablagerung nichtlöslicher Fibrillen, die aus falsch aggregierten Proteinen bestehen, gemein ist. Diese Proteine können sich lokal begrenzt ablagern und dadurch nur wenig Probleme bereiten oder sich sehr ausgedehnt in vielen Organen verbreiten und damit ein schweres Multiorganversagen hervorrufen. Die Amyloidose kann de novo oder sekundär als Folge von verschiedenen Infektionen, Entzündungen oder malignen Tumoren auftreten. Die Diagnose wird durch eine Biopsie des betroffenen Gewebes gestellt; das amyloidogene Protein wird mit einer Vielzahl immunhistologischer und biochemischer Techniken typisiert. Die Behandlung richtet sich nach der Art der Amyloidose.
Amyloidfibrillen bestehen aus normalerweise löslichen, fehlgefalteten Proteinen, die zu Oligomeren und dann zu unlöslichen Fibrillen aggregieren. Eine Vielzahl normaler (Wildtyp) und mutierter Proteine ist für eine solche Fehlfaltung und Aggregation anfällig (amyloidogene Proteine), was zu einer großen Vielfalt an Ursachen und Arten der Amyloidose führt.
Amyloidablagerungen bestehen aus kleinen (etwa 10 nm Durchmesser), unlöslichen Fibrillen, die kongophile Beta-Faltblätter bilden, die durch Röntgen-Beugung identifiziert werden können. Zusätzlich zu den fibrillären Amyloidproteinen enthalten die Ablagerungen Amyloid-P-Komponenten und Glykosaminoglykane.
Amyloidablagerungen färben sich mit Hämatoxylin und Eosin rosa und enthalten Kohlenhydratbestandteile, die sich mit Perjodsäure-Schiff-Farbstoff oder mit Alcian blau färben; am charakteristischsten ist jedoch die apfelgrüne Doppelbrechung unter polarisierter Licht-Mikroskopie nach Kongorotfärbung. Bei einer Autopsie können die betroffenen Organe wächsern wirken.
Damit sich eine Amyloidose entwickelt, muss neben der Produktion amyloidogener Proteine wahrscheinlich auch ein Versagen der normalen Beseitigungsmechanismen für solche fehlgefalteten Proteine vorliegen. Die Amyloidablagerungen selbst sind metabolisch inert, beeinträchtigen die Organfunktion und Struktur aber mechanisch. Einige präfibrilläre Oligomere amyloidogener Proteine haben jedoch eine direkte zelluläre Toxizität, ein wichtiger Bestandteil der Pathogenese der Erkrankung.
Ätiologie der Amyloidose
Bei der systemischen Amyloidose bilden zirkulierende amyloidogen Proteine Ablagerungen in einer Vielzahl von Organen. Zu den wesentlichen systemischen Formen gehören
AL (primäre Amyloidose): verursacht durch eine erworbene Überexpression klonaler Immunglobulin-Leichtketten
AF (familiäre Amyloidose): verursacht durch die Vererbung eines mutierten Gens, das ein zu Fehlfaltungen neigendes Protein enkodiert, meistens Transthyretin (TTR)
ATTRwt (Wildtyp ATTR; früher als senile systemische Amyloidose oder SSA bezeichnet): Verursacht durch Falschfaltung und Aggregation von Wildtyp-TTR
AA (sekundäre Amyloidose): verursacht durch die Aggregation eines Akutphasen-Reaktants, Serum-Amyloid A
Amyloidose, die durch eine Aggregation von Beta-2-Mikroglobulin verursacht wird, kann bei Patienten, die langfristig eine Hämodialyse erhalten, auftreten, aber die Inzidenz ist mit dem Einsatz moderner High-Flow-Dialysemembranen zurückgegangen. Es gibt eine seltene erbliche Form der Beta-2-Mikroglobulin-Amyloidose, die auf einer Mutation des entsprechenden Gens beruht.
Lokalisierte Formen der Amyloidose scheinen durch die lokale Produktion und Ablagerung eines amyloidogenen Proteins (meistens Immunglobulin-Leichtketten) innerhalb des betroffenen Organs verursacht zu werden und nicht durch die Ablagerung zirkulierender Proteine. Häufig beteiligte Lokalisierungen umfassen das Zentralnervensystem (z. B. bei der Alzheimer-Krankheit), die Haut, die oberen oder unteren Atemwege, die Blase und andere Stellen.Häufig beteiligte Seiten sind das ZNS (z. B. bei Alzheimer-Krankheit), Haut, obere oder untere Atemwege, Lungenparenchym, Blase, Augen und Brüste.
AL Amyloidose (primäre Amyloidose)
AL wird durch die Überproduktion einer amyloidogenen Immunglobulin-Leichtkette bei Patienten mit einer monoklonalen Plasmazelle oder einer anderen lymphoproliferativen B-Zellen-Störung verursacht. Leichtketten können auch nicht-fibrilläre Gewebeablagerungen verursachen (d. h. Leichtketten-Speicherkrankheit). In seltenen Fällen bilden Immunglobulin-Schwerketten Amyloidfibrillen (genannt AH Amyloidose).
Bevorzugt von Amyloidablagerungen betroffene Organe sind die Haut, Nerven, das Herz, der Gastrointestinaltrakt (inklusive der Zunge), Nieren, Leber, Milz und auch Blutgefäße. Im Knochenmark liegt in der Regel eine leichte Plasmozytose vor, welche derjenigen beim multiplen Myelom ähnelt, obwohl die meisten Patienten kein echtes multiples Myelom (mit lytischen Knochendefekten, Hyperkalzämie, Ausgüssen der renalen Tubuli und Anämie) haben. Allerdings entwickeln 10–20% aller Patienten mit einem multiplen Myelom eine AL-Amyloidose.
AF-Amyloidose (familiäre Amyloidose)
AF wird durch die Vererbung eines Gens verursacht, das ein mutiertes Protein im Serum enkodiert, welches zur Aggregation neigt; dabei handelt es sich in der Regel um ein Protein, das im Überfluss in der Leber produziert wird.
Zu den Serumproteine, die AF verursachen können, gehören Transthyretin (TTR), Apolipoprotein A-I und Apolipoprotein A-II, Lysozym, Fibrinogen, Gelsolin und Cystatin C. Eine Form, von der angenommen wird, dass sie familiär ist, wird vom Serumprotein Leukozyten-chemotaktischer Faktor 2 (LECT2) verursacht; jedoch wurde keine bestimmte vererbte Genmutation für diesen letzteren Typ eindeutig nachgewiesen.
Die Amyloidose, die durch TTR (ATTR) verursacht wird, ist die häufigste Form von AF. Mehr als 130 Mutationen des TTR-Gens sind mit Amyloidose in Verbindung gebracht worden. Die häufigste Mutation ist V30M und tritt häufig in Portugal, Schweden, Brasilien und Japan auf; zudem liegt eine V122I-Mutation bei etwa 4% der US-amerikanischen und karibischen Schwarzen vor. Die Penetranz der Krankheit und das Erkrankungsalter sind sehr unterschiedlich, jedoch konsistent innerhalb von Familien und ethnischen Gruppen (1).
Die Transthyretin-Amyloidose verursacht periphere sensomotorische Neuropathie und autonome Neuropathie, chronische Nierenerkrankung und Kardiomyopathie. Das Karpaltunnelsyndrom geht gewöhnlich anderen neurologischen Krankheitsmanifestationen voraus. Glaskörperablagerungen können sich aufgrund der Produktion von mutiertem TTR durch das Netzhautepithel entwickeln, oder es kann zu leptomeningealen Ablagerungen kommen, da der Plexus choroideus mutiertes TTR produziert. Wenn die Kardiomyopathie die vorherrschende Manifestation der TTR-Ablagerungen im Herzen ist, wird sie als Transthyretin-Amyloid-Kardiomyopathie (ATTR-CM) bezeichnet.
ATTRwt-Amyloidose (senile systemische Amyloidose)
ATTRwt wird durch Aggregation und Ablagerung von Wildtyp-TTR verursacht, die hauptsächlich auf das Herz abzielen.
ATTRwt wird zunehmend als eine Ursache der infiltrativen Kardiomyopathie bei älteren Männern angesehen. Etwa 16% der Patienten mit Aortenstenose die sich einem Transkatheter-Aortenklappenersatz unterziehen (2) und 13% der Patienten, die wegen Herzinsuffizienz mit erhaltener Auswurffraktion (HFpEF) ins Krankenhaus eingeliefert werden, haben auch eine Transthyretin-Amyloid-Kardiomyopathie, in diesem Fall als wATTR-CM bezeichnet, um die Ablagerung von Wildtyp-TTR im Herzen zu kennzeichnen (3). Weichteilmanifestationen von ATTRwt-Amyloid, einschließlich Karpaltunnelsyndrom, Ruptur der Bizepssehne, Risse der Rotatorenmanschette und Spinalkanalstenose, können der klinischen Ausprägung der infiltrativen Kardiomyopathie um Jahre vorausgehen.
Die genetischen und epigenetischen Faktoren, die zu ATTRwt führen, sind unbekannt. Da ATTRwt und AL-Amyloidose beide eine Kardiomyopathie verursachen können, und weil amyloidogene monoklonale Gammopathien bei Patienten dieser Altersgruppe vorhanden sein können, ist es wichtig, das Amyloid genau zu tippen, damit Patienten mit ATTRwt nicht unangemessen mit einer Chemotherapie behandelt werden (die für AL verwendet wird).
AA Amyloidose (sekundäre Amyloidose)
Diese Form kann sekundär nach verschiedenen Infekten, Entzündungen und malignen Geschehen auftreten und wird durch die Aggregation von Isoformen des Akute-Phase-Proteins Serumamyloid A verursacht.
Häufige ursächliche Infektionen sind
Prädisponierende entzündliche Bedingungen schließen
Vererbte periodische Fiebersyndrome wie familiäres mediterranes Fieber
Castleman-Krankheit
Inflammatorische Zytokine (z. B. Interleukin-1, Tumornekrosefaktor, Interleukin-6), die bei diesen Entzündungen oder ektotypisch von Tumorzellen produziert werden, verursachen eine erhöhte hepatische Synthese von Serumamyloid A (SAA).
AA Amyloidose zeigt sich bevorzugt in der Milz, Leber, den Nieren, den Nebennierendrüsen und den Lymphknoten. Eine Beteiligung des Herzens oder des peripheren und autonomen Nervensystems tritt spät während des Krankheitsverlaufs auf.
Lokalisierte Amyloidose
Die lokalisierte Amyloidose außerhalb des Gehirns wird am häufigsten durch Ablagerungen von klonalen Immunglobulin-Leichtketten verursacht; innerhalb des Gehirns überwiegt das Amyloid-Beta-Protein.
Lokalisierte Amyloid-Ablagerungen betreffen in der Regel die Atemwege und das Lungengewebe, die Blase und den Harnleiter, die Haut, die Brust und die Augen. In seltenen Fällen verursachen andere lokal produzierte Proteine eine Amyloidose, wie etwa Keratin-Isoforme, die lokal Ablagerungen in der Haut bilden können. Klonale Immunglobulin-Leichtketten, die vom mit Schleimhaut assoziiertem lymphatischem Gewebe im gastrointestinalen Trakt, den Atemwegen und der Blase produziert werden, können zu lokalisierter AL in jenen Organen führen.
Amyolid-Beta-Proteinablagerungen im Gehirn tragen zur Alzheimer-Krankheit oder zerebrovaskulären Amyloid-Angiopathie bei. Andere Proteine, die im Zentralnervensystem produziert werden, können Fehlfaltungen bilden, aggregieren und Neuronen schädigen, was zu neurodegenerativen Erkrankungen führt (z. B. Parkinson-Krankheit, Huntington-Krankheit).
Literatur zur Ätiologie
1. Buxbaum JN, Ruberg FL: Transthyretin V122I (pV142I)* cardiac amyloidosis: an age-dependent autosomal dominant cardiomyopathy too common to be overlooked as a cause of significant heart disease in elderly African Americans. Genet Med 19(7):733-742, 2017. doi:10.1038/gim.2016.200
2. Fabbri G, Serenelli M, Cantone A, et al: Transthyretin amyloidosis in aortic stenosis: clinical and therapeutic implications. Eur Heart J Suppl 23(Suppl E):E128-E132, 2021. doi:10.1093/eurheartj/suab107
3. Magdi M, Mostafa MR, Abusnina W, et al: A systematic review and meta-analysis of the prevalence of transthyretin amyloidosis in heart failure with preserved ejection fraction. Am J Cardiovasc Dis 12(3):102-111, 2022. PMID: 35873185
Symptome und Anzeichen von Amyloidose
Die Symptome und Beschwerden der systemischen Amyloidose sind unspezifisch, was oft zur Verzögerung der Diagnosestellung führt. Der Verdacht auf Amyloidose sollte bei Patienten mit einem progressiven Multisystem-Krankheitsprozess verstärkt sein.
Amyloidablagerungen in den Nieren treten typischerweise in der glomerulären Membran auf, was zu Proteinurie führt; in 15% der Fälle sind jedoch die Röhrchen betroffen, was Azotämie mit minimaler Proteinurie verursacht. Diese Prozesse können zum nephrotischen Syndrom mit ausgeprägter Hypoalbuminämie, Ödem und Anasarka oder zu einer Nierenerkrankung im Endstadium voranschreiten.
Eine Leberbeteiligung führt zu einer schmerzlosen Hepatomegalie, die sehr ausgeprägt sein kann. Lebertests deuten typischerweise auf eine intrahepatische Cholestase mit Erhöhung der alkalischen Phosphatase und später des Bilirubin hin, wobei Gelbsucht jedoch selten ist. Gelegentlich entwickelt sich eine portale Hypertonie mit Ösophagusvarizen und Aszites.
Eine Beteiligung der Atemwege und des Larynx führt zu Dyspnoe, Heiserkeit, Keuchen, Hämoptyse oder Atemwegsobstruktion.
Die Infiltration des Herzmuskels verursachtrestriktive Kardiomyopathie, die schließlich zu diastolischer Dysfunktion und Herzversagen führt; ein Herzblock oder eine Herzrhythmusstörung kann auftreten. Hypotonie ist häufig.
Eine periphere Neuropathie mit Parästhesien der Zehen und Finger ist eine häufige Erstmanifestation bei AL- und ATTR-Amyloidosen. Eine autonome Neuropathie kann eine orthostatische Hypotension, erektile Dysfunktion, unnormale Schweißsekretion, Harnverhalt und Störungen der gastrointestinalen Motilität verursachen.
Die zerebrovaskuläre Amyloid-Angiopathie verursacht am häufigsten eine spontane zerebrale Blutung, aber einige Patienten haben kurze, vorübergehende neurologische Symptome.
Gastrointestinales Amyloid verursacht Motilitätsstörungen im Ösophagus, Dünn- und Dickdarm. Eine Atonie des Magens, Malabsorption, Blutungen oder eine Pseudoobstruktion können auftreten. Eine Makroglossie ist häufig bei einer AL-Amyloidose.
Eine Weichteilamyloid-Beteiligung geht charakteristischerweise der klinischen Ausprägung der ATTRwt-Amyloid-Kardiomyopathie voraus. Zu den Manifestationen der Weichteilamyloiderkrankung gehören das Karpaltunnelsyndrom, der Trigger-Finger, die bizipitale Sehnenruptur und die Spinalkanalstenose.

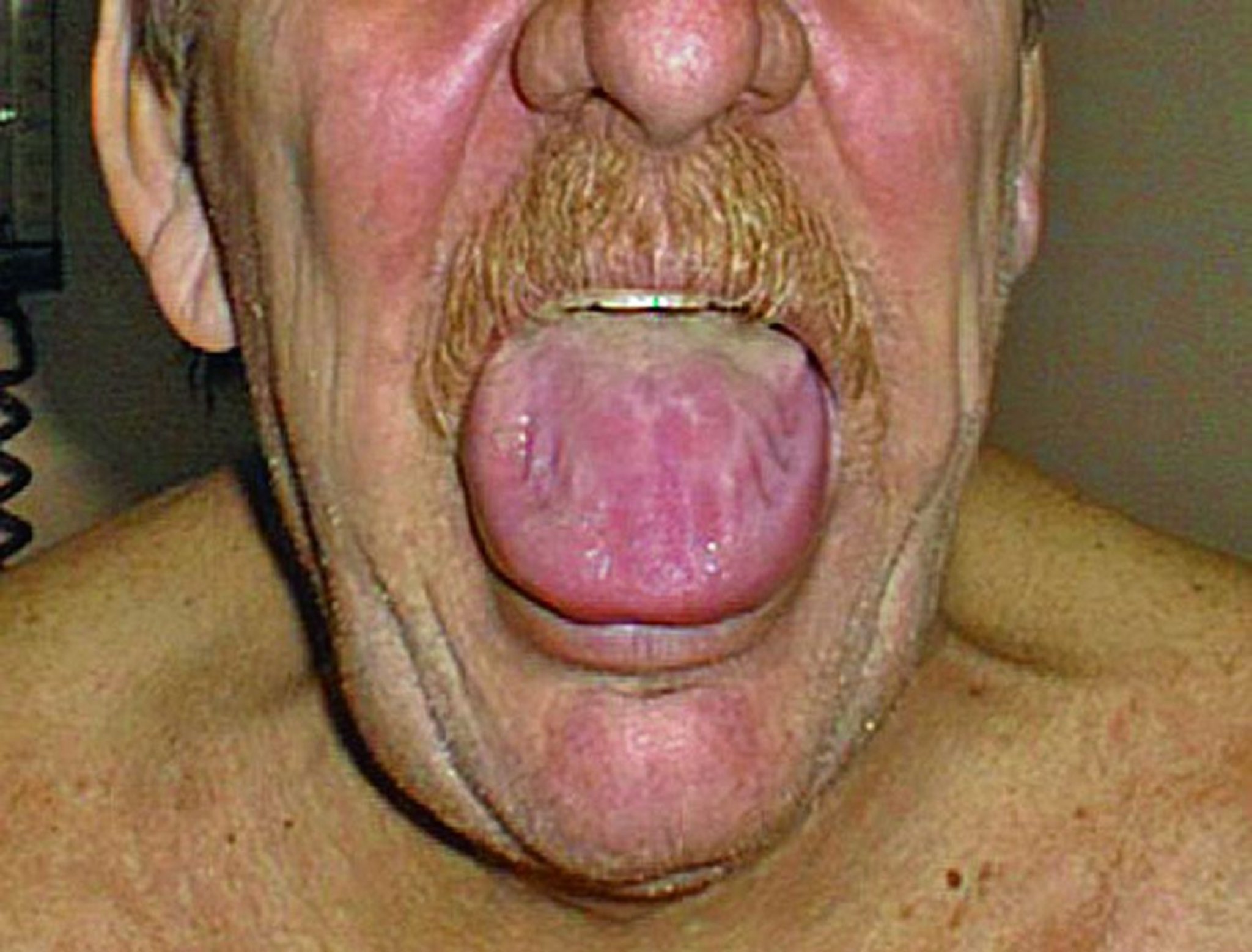
© Springer Science+Business Media
Eine Amyloidose der Schilddrüse kann einen festen, symmetrischen, nicht streckenden Kropf verursachen, der dem bei Hashimoto Thyreoiditis ähnelt. Andere Endokrinopathien können ebenfalls auftreten.
Eine Lungenbeteiligung (meist bei AL-Amyloidose) kann durch fokale Lungenknötchen und -zysten, tracheobronchiale Läsionen, Pleuraergüsse oder diffuse alveolar-septale (interstitielle) Ablagerungen gekennzeichnet sein.
Amyloide Glaskörpertrübungen und bilateral überbackene Pupillenränder entwickeln sich bei mehreren erblichen Amyloidosen.
Weitere Symptome sind Blutergüsse, häufig um die Augen (Waschbäraugen), die durch Amyloidablagerungen in den Blutgefäßen verursacht werden. Amyloidablagerungen verursachen eine Schwächung der Blutgefäße, die nach kleineren Traumata, wie Niesen oder Husten, platzen können.
Diagnose von Amyloidose
Biopsie
Amyloidtypisierung
Testung auf Organbeteiligung
Biopsie
Die Diagnose der Amyloidose wird über die Demonstration fibrillärer Ablagerungen in einem betroffenen Organ gestellt. Bei der Aspiration von subkutanem Bauchfett werden Amyloidablagerungen bei etwa 80% der Patienten mit AL aber weniger als 25% der Patienten mit ATTRwt nachgewiesen (1). Wenn das Ergebnis der Fettbiopsie negativ ist, sollte ein klinisch betroffenes Organ biopsiert werden. Die diagnostische Empfindlichkeit von Nieren- und Herzbiopsien beträgt nahezu 100%, wenn diese Organe klinisch involviert sind. Die Proben werden mit Kongorot angefärbt und mithilfe von polarisierendem Licht auf die charakteristische Doppelbrechung untersucht. Nicht abzweigende 10 nm-Fibrillen können auch durch Elektronenmikroskopie der Biopsieproben aus Herz oder Nieren erkannt werden.
Durch nuklearmedizinische Untersuchungen mit knochenaviden Tracern kann eine ATTR-Amyloid-Kardiomyopathie ohne Herzbiopsie diagnostiziert werden, sofern eine AL-Amyloidose ausgeschlossen ist.
Amyloidtypisierung
Nachdem eine Amyloidose durch Biopsie bestätigt worden ist, wird ihr Typ mit einer Vielzahl von Techniken bestimmt. Für einige Typen der Amyloidose kann die Immunhistochemie oder Immunfluoreszenz diagnostisch sein, aber falsch-positive Typisierungsergebnisse treten auf. Weitere nützliche Techniken umfassen die Gensequenzierung für AF und die biochemische Identifizierung durch Massenspektrometrie um Proteinvarianten in Amyloidablagerungen genau zu identifizieren (die empfindlichste und spezifischste Methode).
Wenn AL vermutet wird, sollten die Patienten auf eine zugrunde liegende Erkrankung der Plasmazellen untersucht werden; dies geschieht mit der quantitativen Messung der freien Immunglobulin-Leichtketten im Serum, dem qualitativen Nachweis monoklonaler Leichtketten mit Immunfixationselektrophorese im Serum oder Urin (Serumprotein-Elektrophorese und Urinprotein-Elektrophorese sind bei Patienten mit AL nicht sensitiv) und einer Knochenmarkbiopsie mit Durchflusszytometrie oder Immunhistochemie, um die Klonalität der Plasmazellen festzustellen.
Patienten mit > 10% klonalen Plasmazellen sollten getestet werden, um zu sehen, ob sie die Kriterien für das multiple Myelom erfüllen, einschließlich des Screenings auf lytische Knochenläsionen, Anämie, Niereninsuffizienz und Hyperkalzämie.
Organbeteiligung
Die Patienten werden auf eine Organbeteiligung untersucht, beginnend mit nichtinvasiven Tests:
Nieren: Urinanalyse; Messung von BUN im Serum, Kreatinin und Albumin; geschätzte glomeruläre Filtrationsrate (eGFR); und 24-Stunden-Urinsammlung für Proteinelektrophorese (UPEP)
Leber: Bestimmung der Leber(funktions)parameter
Lungen: Thoraxröntgen-, Thorax-CT- und Lungenfunktionstests
Herz: EKG und Messung von Biomarker wie natriuretischem Peptid (BNP) oder N-terminalen pro-BNP (NT-proBNP) und Troponin
Eine Herzbeteiligung kann durch eine niedrige Spannung im EKG (verursacht durch eine verdickte Herzkammer) und/oder Rhythmusstörungen angedeutet werden. Besteht aufgrund der Symptome der Verdacht auf eine kardiale Beteiligung, wird zusätzlich zu den EKG-Befunden und den kardialen Biomarkern eine Echokardiographie durchgeführt, um die diastolische Relaxation und die globale Längsdehnung (ein Maß für die linksventrikuläre systolische Funktion) zu messen und auf eine biventrikuläre Hypertrophie zu untersuchen. In Zweifelsfällen kann ein Herz-MRT durchgeführt werden, um eine anhaltende subendokardiale Gadoliniumsteigerung nachzuweisen, ein charakteristischer Befund. Die Technetium-Pyrophosphat-Nuklearszintigraphie verbessert die Erkennung der ATTR-Amyloid-Herzerkrankung und kann die Notwendigkeit von Herzbiopsien vermeiden, sofern Bluttests eine AL-Amyloidose ausschließen (2, 3).
Literatur zur Diagnose
1. Aimo A, Emdin M, Musetti V, et al: Abdominal Fat Biopsy for the Diagnosis of Cardiac Amyloidosis. JACC Case Rep 2(8):1182-1185, 2020. doi:10.1016/j.jaccas.2020.05.062
2. Gillmore JD, Maurer MS, Falk RH, et al: Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation 133(24):2404–2412, 2016.
3. Maurer MS, Bokhari S, Damy T, et al: Expert consensus recommendations for the suspicion and diagnosis of transthyretin cardiac amyloidosis. Circ Heart Fail 12(9):e006075, 2019.
Behandlung der Amyloidose
Unterstützende Behandlung
Typspezifische Behandlung
Gibt es spezielle Behandlungen für die meisten Formen der Amyloidose, obwohl sich einige Behandlungen in der Prüfungsphase befinden. Bei allen Formen der systemischen Amyloidose können supportive Pflegemaßnahmen dabei helfen, die Symptome zu lindern und die Lebensqualität zu erhöhen.
Unterstützende Behandlung
Die supportiven Pflegemaßnahmen sind auf das betroffene Organsystem gerichtet:
Renal: Patienten mit nephrotischem Syndrom und Ödem sollten mit Salz und Flüssigkeitsrestriktion sowie Loop-Diuretika behandelt werden; aufgrund des anhaltenden Proteinverlusts sollte die Proteinzufuhr nicht eingeschränkt werden. Eine Nierentransplantation stellt eine Option dar, wenn der zugrunde liegende Krankheitsprozess kontrolliert wird; sie kann zu einem langfristigen Überleben führen, das mit demjenigen bei anderen Nierenkrankheiten vergleichbar ist.
Kardial: Patienten mit Kardiomyopathie sollten mit Salz und Flüssigkeitsrestriktion sowie Loop-Diuretika behandelt werden. Andere Medikamente gegen Herzversagen, einschließlich Digoxin, Angiotensin-konvertierendes Enzym-Inhibitoren, Kalzium-Antagonisten und Beta-Blocker werden schlecht vertragen und sind kontraindiziert. Bei sorgfältig ausgewählten Patienten mit einer AL- oder ATTR-Amyloidose und massiver Herzbeteiligung war eine Herztransplantation erfolgreich. Um ein Wiederauftreten des transplantierten Herzens zu verhindern, müssen Patienten mit AL-Amyloidose eine aggressive Chemotherapie gegen die klonale Plasmazellerkrankung erhalten. Patienten mit symptomatischer ATTR-Amyloid-Polyneuropathie oder Kardiomyopathie sollten für Anti-TTR-Therapien in Betracht gezogen werden.
Magen-Darm: Patienten mit Diarrhö können von Loperamid profitieren. Diejenigen mit frühem Sättigungsgefühl und Magenentleerung können von Metoclopramid profitieren.
Nervensystem: Bei Patienten mit peripherer Neuropathie kann Gabapentin oder Pregabalin die Schmerzen lindern.
Eine orthostatische Hypotonie verbessert sich oft mit hohen Dosen von Midodrin; dieses Medikament kann bei älteren Männern Harnverhalt verursachen, aber medikamentöse Komplikation von Hypertonie in Rückenlage ist in dieser Population selten ein Problem. Stützstrümpfe können ebenfalls helfen und Fludrocortison kann bei Patienten ohne peripheres Ödem, Anasarka oder Herzversagen verwendet werden. Bei Patienten mit refraktärer orthostatischer Hypotonie können Midodrin, Fludrocortison oder Droxidopa eingesetzt werden.
AL-Amyloidose
Bei AL-Amyloidose
Rasche Einleitung der Antiplasma-Zelltherapie wichtig, um die Organfunktion zu erhalten und das Leben zu verlängern.
Die meisten Medikamente, die gegen das multiple Myelom verwendet werden sind bei AL-Amyloidose eingesetzt worden; die Auswahl des Medikaments, seine Dosis und der Zeitplan müssen häufig modifiziert werden, wenn die Organfunktion beeinträchtigt ist.
Eine Chemotherapie, die ein Alkylierungsmittel (z. B. Melphalan, Cyclophosphamid) einsetzt, in Kombination mit Kortikosteroiden war die erste Behandlung, die sich als nützlich erwiesen hat. Hochdosiertes Melphalan IV kombiniert mit einer autologen Stammzelltransplantation kann bei ausgewählten Patienten sehr effektiv sein (1).
Proteasom-Inhibitoren (z. B. Bortezomib) und Immunmodulatoren (z. B. Lenalidomid) können auch wirksam sein. Eine Studie mit dem monoklonalen Antikörper Daratumumab plus Cyclophosphamid, Bortezomib und Dexamethason bei Patienten mit neu diagnostizierter AL-Amyloidose (unter Ausschluss von Patienten mit Herzinsuffizienz der der NYHA-Klassen III und IV, N-terminalem Pro-B-Typ-Natriuretik-Protein [NTproBNP] > 8,500 pg/ml [> 1005 pmol/l] und eGFR < 20 ml/Minute/m2) zeigte eine beispiellos hohe Rate an hämatologischem Ansprechen (2). Das hämatologische Ansprechen basiert auf dem Gehalt an monoklonalem Protein in Serum und Urin, der durch Immunofixationselektrophorese bestimmt wird, sowie auf dem Gehalt an leichten Ketten im Serum mit dem Kappa/Lambda-Verhältnis. Es fehlen jedoch Daten zum Langzeitüberleben.
Alle verfügbaren Behandlungen richten sich gegen klonale B-Zellen oder Plasmazellen bei AL-Amyloidose. Studien zu Antifibrillen-Antikörpern, wie Birtamimab und CAEL-101, sind im Gange (3).
Lokalisierte AL-Amyloidose kann mit einer niedrig dosierten externen Strahlentherapie behandelt werden, da Plasmazellen sehr strahlenempfindlich sind.
ATTR-Amyloidose
Für ATTR-Amyloidose:
Lebertransplantation
Behandlungsstabilisierende Medikamente
Gen-Silencing-Arzneimittel
Eine Lebertransplantation, bei der der primäre Ort der Synthese des mutierten Proteins durch ein neues Organ ersetzt wird, das normales TTR produziert, kann bei bestimmten TTR-Mutationen wirksam sein, wenn sie zu Beginn der Krankheit durchgeführt wird (frühe Neuropathie und keine Herzbeteiligung). Eine Transplantation im späteren Verlauf der Krankheit führt oft zu einer progressiven Amyloid-Kardiomyopathie und Neuropathie aufgrund der Fehlfaltung und Ablagerung von Wildtyp-TTR-Protein auf vorhandenen Amyloid-Ablagerungen.
Mehrere Medikamente stabilisieren nachweislich die im Plasma zirkulierenden TTR-Tetramere, hemmen eine Fehlfaltung und Fibrillenbildung der TTR und verlangsamen effektiv das Fortschreiten der neurologischen Erkrankung unter Wahrung der Lebensqualität. Zu diesen TTR-Stabilisatoren gehören Diflunisal, ein weit verbreitetes generisches entzündungshemmendes Medikament, und Tafamidis (4, 5).
TTR-Gen-Schalldämpfer mit Hilfe von Anti-Sense-RNA oder RNA-Interferenz zur Blockierung der Translation der TTR-mRNA senkt die TTR-Serumspiegel wirksam, verbessert die neurologischen Ergebnisse bei etwa 50% der Patienten und scheint bei einigen Patienten in der Lage zu sein, verletzte Nerven zu reparieren (6, 7). Die Gen-Silencing-Arzneimittel, Patisiran, Inotersen und Vutrisiran sind verfügbar.
Eine Studie mit Vutrisiran, einem Gen-Silencer der zweiten Generation, zeigte verbesserte funktionelle Ergebnisse bei Patienten mit familiärer Amyloid-Polyneuropathie (8). Vorläufige Daten aus einer anderen Studie deuten darauf hin, dass Gen-Silencer bei der Behandlung der Kardiomyopathie von Patienten mit ATTR-Amyloidose wirksam sein könnten (9).
ATTRwt-Amyloidose
ABei TTRwt-Amyloidose
Behandlungsstabilisierende Medikamente
Die TTR-Stabilisierung mit Tafamidis bei Patienten mit ATTR oder ATTRwt hat gezeigt, dass Amyloid-Kardiomyopathie die Gesamtmortalität und kardiovaskuläre Krankenhauseinweisungen verringert (5). Derzeit werden klinische Studien durchgeführt, in denen die Wirkung von TTR-Gen-Silencern auf die Kardiomyopathie bei Patienten mit ATTRwt-Amyloidose sowie auf die Kardiomyopathie bei Patienten mit ATTR-Amyloidose, die durch das mutierte Protein gekennzeichnet ist, untersucht wird (10).
Im Gegensatz zur erblichen ATTR-Amyloidose ist eine Lebertransplantation bei Patienten mit ATTRwt nicht wirksam, da das amyloidogene Protein eine strukturell normale TTR ist.
AA-Amyloidose
Bei AA-Amyloidose, die durch familiäres Mittelmeerfieber verursacht wird, ist orales Colchicin wirksam.
Bei anderen AA-Typen wird die Behandlung auf die zugrunde liegende Infektion, entzündliche Erkrankung oder Krebs gerichtet.
Colchicin oder Anti-IL1-, Anti-IL6- oder Anti-TNF-Medikamente können eingesetzt werden, um die Zytokin-Signalübertragung zu unterbrechen und den Entzündungsprozess, der die hepatische Produktion von Serum-Amyloid A (SAA) antreibt, zu verringern.
Literatur zur Behandlung
1. Sanchorawala V, Sun F, Quillen K, et al: Long-term outcome of patients with AL amyloidosis treated with high-dose melphalan and stem cell transplantation: 20-year experience. Blood 126: 2345–2347, 2015. doi: 10.1182/blood-2015-08-662726
2. Kastritis E, Palladini G, Minnema MC, et al: Daratumumab-Based Treatment for Immunoglobulin Light-Chain Amyloidosis. N Engl J Med 385(1):46-58, 2021. doi:10.1056/NEJMoa2028631
3. Quarta CC, Fontana M, Damy T, et al: Changing paradigm in the treatment of amyloidosis: From disease-modifying drugs to anti-fibril therapy. Front Cardiovasc Med 9:1073503, 2022. doi:10.3389/fcvm.2022.1073503
4. Berk JL, Suhr OB, Obici L, et al: Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA 310: 2658–2667, 2013. doi: 10.1001/jama.2013.283815
5. Maurer MS, Schwartz JH, Gundapaneni B, et al: Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med 379:1007–1016, 2018.
6. Adams D, Gonzalez-Duarte A, O'Riordan WD, et al: Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med 379:11–21, 2018.
7. Benson MD, Waddington-Cruz M, Berk JL, et al: Inotersen treatment for patients with transthyretin amyloidosis. N Engl J Med 379:22–31, 2018.
8. Adams D, Tournev IL, Taylor MS, et al: Efficacy and safety of vutrisiran for patients with hereditary transthyretin-mediated amyloidosis with polyneuropathy: a randomized clinical trial. Amyloid 30(1):1-9, 2023. doi:10.1080/13506129.2022.2091985
9. Maurer MS, Fontanta MA, Berk JL, et al: Primary results from APOLLO-B, a phase 3 study of patisiran in patients with transthyretin-mediated amyloidosis with cardiomyopathy. Abstract presented at International Symposium of Amyloidosis, September 2022,Heidelberg Germany.
10. Writing Committee, Kittleson MM, Ruberg FL, et al: 2023 ACC Expert Consensus Decision Pathway on Comprehensive Multidisciplinary Care for the Patient With Cardiac Amyloidosis: A Report of the American College of Cardiology Solution Set Oversight Committee [published correction appears in J Am Coll Cardiol 81(11):1135, 2023]. J Am Coll Cardiol 81(11):1076-1126, 2023. doi:10.1016/j.jacc.2022.11.022
Prognose der Amyloidose
Die Prognose ist von der Art der Amyloidose und von den befallenen Organsystemen abhängig, wobei viele Patienten jedoch mit angemessener krankheitsspezifischer und supportiver Pflege eine exzellente Lebenserwartung haben.
Eine durch schwere Kardiomyopathie verkomplizierte AL hat die schlechteste Prognose mit einem mittleren Überleben von < 1 Jahr. Eine unbehandelte ATTR-Amyloidose entwickelt sich in der Regel innerhalb von 5 bis 15 Jahren zu einer kardialen oder neurologischen Krankheit im Endstadium. Es wurde einmal angenommen, dass ATTRwt das langsamste Fortschreiten einer systemischen Amyloidose am Herzen hat; Patienten mit ATTRwt kommen jedoch innerhalb eines Medians von 4 Jahren nach der Biopsiediagnose zu symptomatischer Herzinsuffizienz und Tod.
Die Prognose der AA-Amyloidose hängt größtenteils von der Wirksamkeit der Behandlung der zugrunde liegenden infektiösen, entzündlichen oder malignen Erkrankung ab.
Wichtige Punkte
Amyloidose ist eine Gruppe von Erkrankungen, bei denen bestimmte fehlgefaltete Proteine zu unlöslichen Fibrillen aggregieren, die in Organen abgelagert werden, was zu Funktionsstörungen führt.
Viele verschiedene Proteine neigen zu Fehlfaltungen; einige dieser Proteine werden durch einen Gendefekt oder durch bestimmte Krankheitszustände produziert, während andere Immunglobulin-Lichtketten einbeziehen, die durch monoklonale Plasmazellen oder andere lymphoproliferative Störungen der B-Zellen produziert werden.
Das amyloidogene Protein bestimmt den Amyloid-Typ und den klinischen Verlauf der Erkrankung, obwohl sich die klinischen Manifestationen der verschiedenen Typen überlappen können.
Viele Organe können betroffen sein, aber eine Herzbeteiligung führt zu einer besonders schlechten Prognose; Amyloidkardiomyopathie führt typischerweise zu diastolischer Dysfunktion, Herzversagen und Herzblock oder Herzrhythmusstörungen.
Die Diagnose wird durch Biopsie gestellt; der Typ der Amyloidose wird durch eine Vielzahl immunologischer, genetischer und biochemischer Tests bestimmt. Massenspektrometrie ist die empfindlichste und spezifischste Methode für die Amyloidtypisierung.
Eine angemessene supportive Pflege hilft dabei, die Symptome zu lindern und die Lebensqualität zu erhöhen; eine Organtransplantation kann ausgewählten Patienten helfen.
Behandeln Sie den zugrunde liegenden Prozess; bei AL-Amyloidose aufgrund von Plasmazellen oder lymphoproliferativen Störungen kann die Chemotherapie hochwirksam sein; bei sekundärer AA-Amyloidose können antiinfektiöse und entzündungshemmende Medikamente helfen.
Bei der erblichen ATTR-Amyloidose hemmen niedermolekulare Stabilisator-Therapeutika und Gene-Silencing-Medikamente die neurologische Verschlechterung oder machen sie potenziell rückgängig. Bei Patienten mit Amyloid-Kardiomyopathie (ATTR oder ATTRwt) verringert Tafamidis die Gesamtmortalität und kardiovaskuläre Krankenhauseinweisungen.