




A vasculite é uma inflamação dos vasos sanguíneos, frequentemente com isquemia, necrose e inflamações de órgãos. A vasculite pode afetar qualquer vaso sanguíneo—artérias, arteríolas, veias, vênulas ou vasos capilares. As manifestações clínicas de distúrbios vasculíticos específicos são diversas e dependem do tamanho e local dos vasos envolvidos, extensão do envolvimento dos órgãos e o grau e padrão da inflamação extravascular.
Etiologia da vasculite
Vasculites podem ser
Primária
Secundária
Vasculite primária não tem causa conhecida.
Vasculite secundária pode ser desencadeada por uma infecção, fármaco ou toxina ou pode ocorrer como parte de outro distúrbio inflamatório ou câncer.
Fisiopatologia da vasculite
A descrição histológica de um vaso afetado deve incluir o seguinte:
Uma descrição do dano na parede do vaso (p. ex., tipo e local do infiltrado inflamatório, extensão e tipo de dano, presença ou ausência de necrose fibrinoide)
Uma descrição das respostas cicatriciais (p. ex., hipertrofia da íntima e fibrose)
Certas características (p. ex., células inflamatórias predominantes, local da inflamação) sugerem processos vasculíticos específicos e podem auxiliar o diagnóstico. Por exemplo, em muitas lesões agudas, as células inflamatórias predominantes são os leucócitos polimorfonucleares; nas lesões crônicas, os linfócitos predominam.
A inflamação pode ser segmentar ou envolver todo o vaso. Nos locais da inflamação, ocorrem graus variados de inflamação celular e necrose ou cicatrização, em uma ou mais camadas da parede do vaso. A inflamação na média de uma artéria muscular tende a destruir a lâmina elástica interna. Algumas formas de vasculite são caracterizadas por células gigantes na parede do vaso. Em algumas vasculites, como a granulomatose com poliangiite ou a doença de Kawasaki, a inflamação do vaso (vasculite verdadeira) é apenas parte da fisiopatologia e há predomínio da inflamação parenquimatosa em um padrão característico comprometendo determinados órgãos.
“Vasculite leucocitoclástica” é um termo histopatológico utilizado para descrever descobertas de vasculites em pequenos vasos. Ele se refere à ruptura das células com inflamação que deixa pequenos fragmentos nucleares (resíduos nucleares) dentro e em volta dos vasos. A inflamação é transmural e não granulomatosa. Os leucócitos polimorfonucleares predominam no início; os linfócitos predominam posteriormente. A resolução da inflamação tende a se resolver com fibrose e hipertrofia da íntima. A hipertrofia da íntima ou a formação secundária de um coágulo pode estreitar o lúmen do vaso e causar isquemia ou necrose tecidual.
Classificação da vasculite
Os distúrbios vasculíticos podem ser classificados de acordo com o tamanho dos vasos envolvidos. Entretanto, frequentemente há sobreposições substanciais.
Classificação dos distúrbios vasculíticos
Tamanho dos vasos predominantemente comprometidos | Doenças | Sinais e sintomas |
|---|---|---|
Grande | Claudicação do membro Medições da pressão arterial desiguais ou pulso desigual, força/ausência de pulso nos membros Sintomas isquêmicos do sistema nervoso central (p. ex., acidente vascular encefálico) | |
Médio | Sintomas de infarto tecidual nos órgãos afetados, como
Sinais e sintomas na doença de Kawasaki: febre, exantema, linfadenopatia, conjuntivite e aneurismas das artérias coronárias | |
Pequeno | Granulomatose eosinofílica com poliangiite Vasculite crioglobulinêmica Vasculite associada à imunoglobulina A (anteriormente chamada púrpura de Henoch-Schönlein) | Sintomas de infarto tecidual nos órgãos afetados similares àqueles para vasos de tamanho médio, exceto lesões cutâneas que têm maior probabilidade de serem purpúricas Rim: glomerulonefrite (geralmente assintomática) |
Sinais e sintomas da vasculite
O tamanho dos vasos afetados ajuda a determinar a apresentação clínica.
Independentemente do tamanho dos vasos envolvidos, os pacientes podem apresentar sinais e sintomas de inflamação sistêmica (p. ex., febre, suor noturno, fatiga, anorexia, perda ponderal, artralgia e artrite). Algumas manifestações são letais ou ameaçam órgãos e requerem tratamento imediato:
Vasculites de tamanho pequeno e médio frequentemente se manifestam com lesões cutâneas como púrpura palpável, urticária, úlceras, livedo reticular e nódulos.

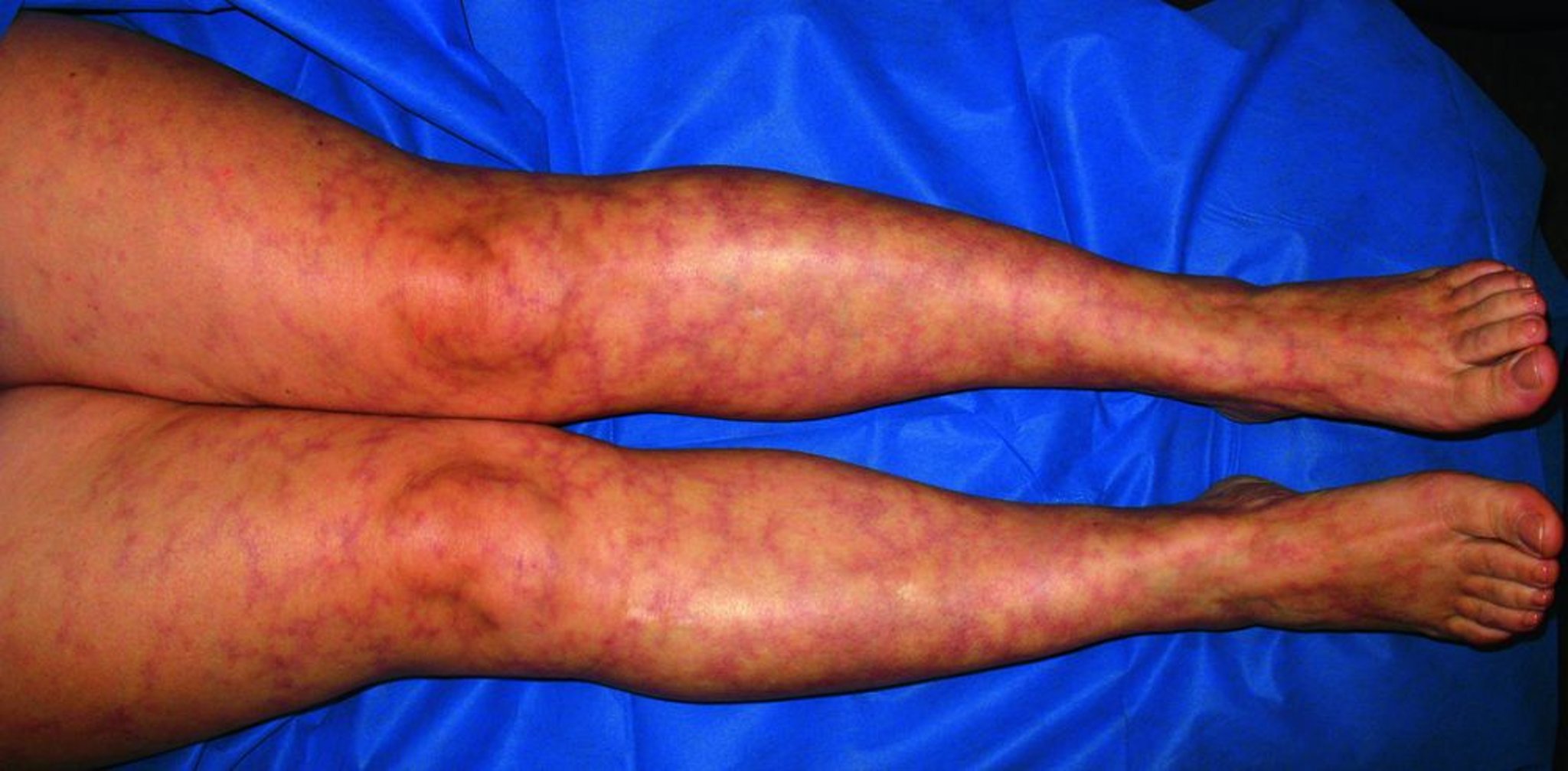
© Springer Science+Business Media

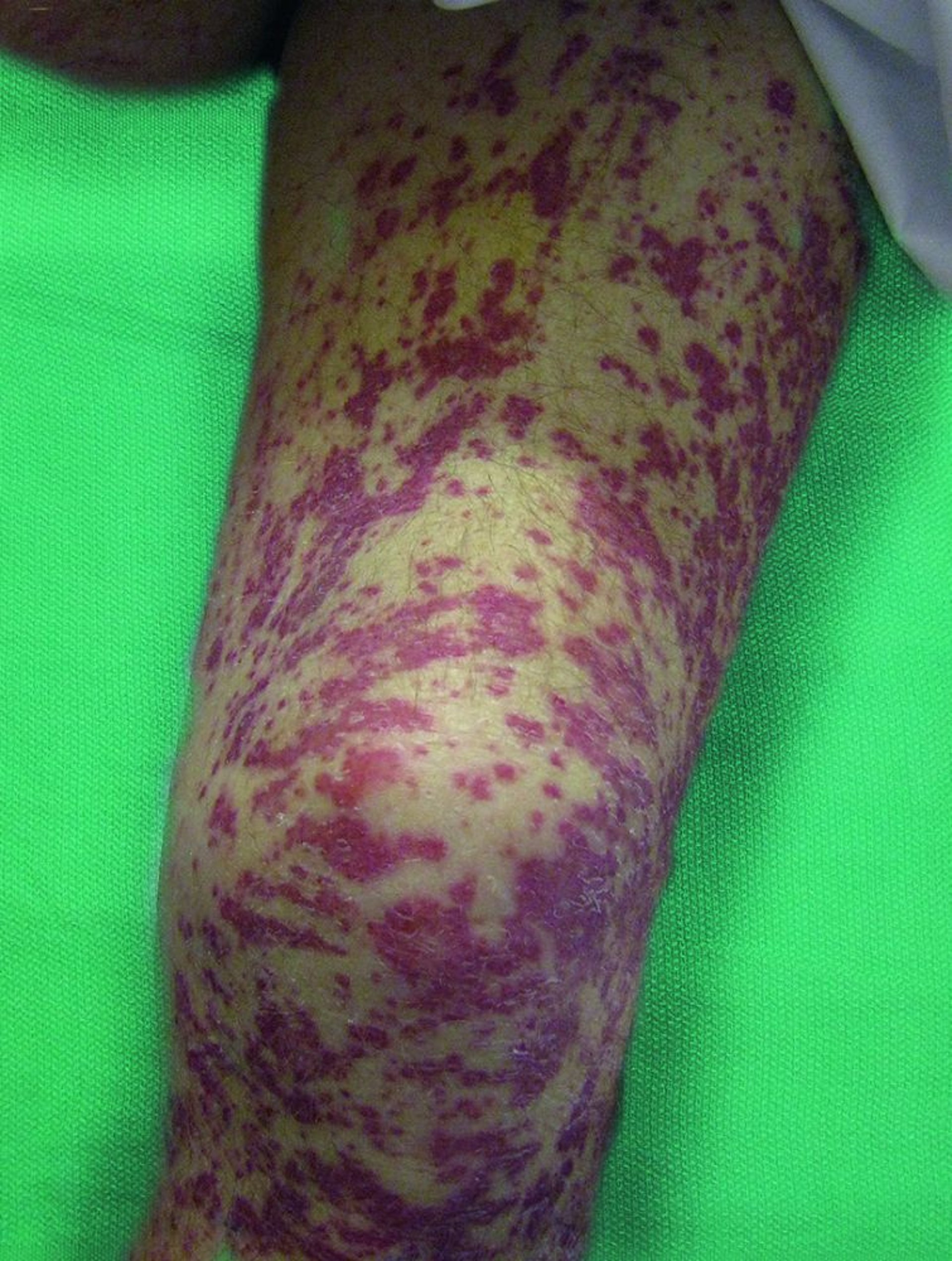
© Springer Science+Business Media
Diagnóstico da vasculite
Avaliação clínica
Exames laboratoriais básicos para detectar inflamação ou disfunção orgânica [p. ex., hemograma completo, velocidade de sedimentação de eritrócitos ou proteína C reativa, albumina sérica e níveis de proteína totais, aspartato aminotransferase (AST) e alanina aminotransferase (ALT), nitrogênio da ureia sanguínea e creatinina séricos eurinálise] e para estagiar o processo da doença
Exames laboratoriais para ajudar a determinar o tipo de vasculite [p. ex., anticorpos anticitoplasma de antineutrófilos (ANCA)] segundo indicação clínica
Estudos laboratoriais e de imagem que podem ajudar a determinar a causa da vasculite (p. ex., crioglobulinas, teste de antígeno de superfície da hepatite B, testes de anticorpos de superfície de núcleo e hepatite B e teste de anticorpos do vírus da hepatite C, hemoculturas) e extensão do envolvimento de órgãos
Biópsia
A vasculite sistêmica é suspeitada nos pacientes com os seguintes sintomas:
Sinais ou sintomas sugestivos de vasculite (p. ex., cefaleia temporal e claudicação maxilar sugerindo arterite de células gigantes)
Manifestações isquêmicas (p. ex., acidente vascular encefálico isquêmico, claudicação do membro e isquemia mesentérica) desproporcionais em relação aos fatores de risco do paciente para aterosclerose
Combinações inexplicáveis de sinais e sintomas em mais de um sistema de órgãos compatíveis com vasculite (p. ex., hipertensão, mialgia, hemoptise), particularmente quando existem sinais e sintomas de doença sistêmica
Os distúrbios vasculíticos primários são diagnosticados com base na presença de sintomas característicos, achados físicos, resultados de exames laboratoriais e exclusão de outras causas (isto é, vasculite secundária). Sempre que possível fazer exames citopatológicos que possam corroborar o diagnóstico de determinada vasculite (ver tabela Dicas histológicas para o diagnóstico de distúrbios vasculíticos). Os achados clínicos determinam o diagnóstico diferencial e, assim, direcionam os exames laboratoriais.
A maioria dos exames laboratoriais de rotina produz resultados inespecíficos e deve-se interpretá-los no contexto de todas as manifestações clínicas. Mas esses resultados muitas vezes podem ajudar a corroborar o diagnóstico, determinar a localização e o grau de comprometimento dos órgãos ou sugerir outros diagnósticos. Os exames geralmente incluem: hemograma, VHS ou proteína C-reativa, albumina sérica e proteína total, AST e ALT. Frequentemente os pacientes apresentam VHS ou proteína C reativa elevados, anemia devido a inflamação crônica, aumento de plaquetas e albumina sérica baixa. Urina recém-coletada deve ser testada para contagem do número de eritrócitos, cilindros hemáticos e proteína para identificar o envolvimento renal. Os níveis de creatinina sérica devem ser checados e monitorados. Leucopenia e trombocitopenia não são típicas da vasculite primária e sugerem um diagnóstico alternativo.
A detecção do ANCA (antineutrophil cytoplasmic antibodies) pode corroborar o diagnóstico de granulomatose com poliangiite (GPA), granulomatose eosinofílica com poliangiite (GEPA) ou poliangiite microscópica (chamadas coletivamente vasculites associadas ao ANCA). Os testes padronizados para ANCA incluem coloração de imunofluorescência e ensaio imunoenzimático (ELISA). A coloração de imunofluorescência nos neutrófilos fixados em etanol pode detectar o padrão citoplasmático do c-ANCA ou o padrão perinuclear do p-ANCA. Então ELISA é utilizado para verificar anticorpos específicos para os principais autoantígenos: proteinase-3 (PR3), que produz o padrão de coloração c-ANCA ou mieloperoxidase (MPO), que produz o padrão de coloração p-ANCA visto em neutrófilos fixados em etanol. Como as vasculites associadas ao ANCA são raras, e o teste do ANCA não é completamente específico, os testes de ANCA devem ser feitos somente quando a probabilidade de vasculite associada ao ANCA antes do teste for moderadamente alta. Teste ANCA positivo pode ocorrer em infecções que podem causar vasculite secundária, incluindo endocardite.
Outros exames laboratoriais úteis são sorologias para hepatite B e C, eletroforese de proteínas séricas e urinárias, anticorpos antinucleares e o perfil de antígenos nucleares antiextraíveis, testar a presença de crioglobulinemia e os níveis do complemento. Os níveis de complemento podem ser baixos em caso de vasculite viral, vasculite crioglobulinêmica, distúrbios linfoproliferativos ou vasculites secundárias a outras doenças autoimunes.
Exames adicionais são determinados de acordo com os achados clínicos. Se indicada com base nos resultados clínicos, uma radiografia do tórax deve ser feita para verificar infiltrados pulmonares, mas uma tomografia torácica de alta resolução sem contraste pode ser necessária para verificar achados sutis, tais como pequenos nódulos ou cavidades. Infiltrados difusos bilaterais sugerem uma possível hemorragia alveolar, o que requer um rápido diagnóstico e tratamento. Outros testes de imagem podem ser necessários. Por exemplo, angiografia por ressonância magnética dos grandes vasos sanguíneos e da aorta é importante para diagnosticar e monitorar em caso de suspeita do comprometimento desses vasos. Se os sintomas e exames sugerem uma neuropatia, a eletromiografia pode ser útil.
Como distúrbios vasculíticos são raros e têm sérios efeitos adversos, a biópsia tecidual deve ser obtida para confirmar o diagnóstico sempre que possível. Os resultados clínicos sugerem o melhor local para a biópsia. Os resultados da biópsia têm maior probabilidade de serem positivos se as amostras forem de tecido pulmonar, pele ou tecido renal atingidos. Biopsias cegas dos órgãos sem manifestações clínicas ou sugestão laboratorial de comprometimento têm baixa probabilidade de resultados positivos.
Tratamento da vasculite
Induzir a remissão da vasculite potencialmente fatal ou que ameaça órgãos vitais com corticoides, muitas vezes associados a ciclofosfamida ou rituximabe
Indução da remissão para formas menos graves de vasculite com corticoides e imunossupressores menos potentes (p. ex., metotrexato, azatioprina e micofenolato mofetila) ou rituximabe
Manutenção da remissão com metotrexato, azatioprina ou rituximabe, além do desmame dos corticoides
O tratamento da vasculite depende da etiologia, do tipo de vasculite, da extensão e da gravidade da doença. No caso de vasculite secundária, a remoção da causa (p. ex., infecção, fármacos, câncer) costuma ajudar.
Na vasculite primária, o objetivo do tratamento é induzir e manter a remissão. A remissão é induzida utilizando imunossupressores citotóxicos e altas doses de corticoides, geralmente por 3 a 6 meses, até a remissão ocorrer ou até a atividade da doença ser reduzida a um nível aceitável. A duração da remissão é difícil de prever e pode depender do tipo de vasculite. Para muitos pacientes, manter a remissão exige a continuação da terapia imunossupressora com ou sem dose baixa de corticoides. Durante esse período, o objetivo é eliminar os corticoides ou reduzir a dose e utilizar imunossupressores alternativos menos tóxicos conforme necessário.
Todos os pacientes tratados com imunossupressores devem ser monitorados para infecções oportunistas e outras. Deve-se considerar testar a tuberculose e a hepatite B, que podem ser reativadas por alguns tratamentos imunossupressores. A profilaxia contra o deve ser considerada para os pacientes que recebem terapia imunossupressora potente ou prolongada.
Indução da remissão
Para as formas menos graves de vasculite, pode-se utilizar baixas doses de corticoides e imunossupressores menos potentes (p. ex., metotrexato, azatioprina e micofenolato de mofetila).
A vasculite grave, rapidamente progressiva e que causa risco ao órgão e à vida do paciente (p. ex., que causa hemorragia alveolar, glomerulonefrite progressiva ou isquemia mesentérica) é uma emergência médica que requer internação hospitalar e tratamento imediato. O tratamento tipicamente consiste em:
Corticoides: altas doses de corticoides (também chamado de pulso de corticoides) também são prescritas. Deve-se individualizar as doses e os fármacos específicos. Como exemplo, utilizar metilprednisolona 15 mg/kg ou 1 g IV uma vez ao dia durante 3 dias, seguida de prednisona ou metilprednisolona 1 mg/kg por via oral (ou, se o paciente estiver internado, algumas vezes IV), uma vez ao dia durante cerca de 4 semanas. Reduz-se então a dose lentamente, como tolerado, até que o fármaco seja interrompido. Pode ser necessária uma mudança nessa escala de redução do fármaco, caso o paciente não apresente melhora, ou em caso de recidiva.
Ciclofosfamida: uma dose de 2 mg/kg por via oral uma vez ao dia é geralmente recomendada por pelo menos 3 meses ou até a remissão. A contagem de leucócitos deve ser monitorada e a dose ajustada para evitar leucopenia. (A contagem de leucócitos deve ser mantida em > 3500/microL [> 3,5 x 109/L].) Alternativamente, às vezes utiliza-se um esquema com ciclofosfamida IV de 0,5 a 1 g/m 2 em intervalos de 2 a 4 semanas. A dose deve ser reduzida em pacientes com insuficiência renal significativa e contagens de leucócitos devem ser monitoradas com frequência. Os pacientes tomando altas doses crônicas de corticoides, especialmente com ciclofosfamida, também devem receber tratamento profilático contra .
Mesna:mesna é misturado com ciclofosfamida IV para se ligar a acroleína, produto da degradação da ciclofosfamida tóxico para o epitélio vesical e pode causar cistite hemorrágica e, às vezes, carcinoma de células transicionais da bexiga. O uso prolongado da ciclofosfamida aumenta o risco de câncer de bexiga. Um miligrama de mesna é utilizado para cada miligrama de ciclofosfamida. A recorrência de hematúria, especialmente sem cilindros e eritrócitos dismórficos, sugere uma avaliação urológica. A citoscopia e exames de imagem devem ser feitos para excluir câncer.
Rituximabe: foi demonstrado que o rituximabe, um anticorpo monoclonal anti-CD20 que depleta células B, não é inferior à ciclofosfamida na indução da remissão da vasculite grave associada ao ANCA. Rituximabe é administrado a 375 mg/m2 IV uma vez por semana durante 4 semanas. Um esquema alternativo amplamente utilizado é o de duas infusões de 1000 mg administradas com 2 semanas de intervalo. Os pacientes também devem receber tratamento profilático contra .
As terapias de depleção de linfócitos B diminuirão acentuadamente a resposta às vacinas por meses depois da administração.
Manutenção da remissão
Os corticoides são diminuídos para zero ou para a menor dose que possa manter a remissão. Para algumas formas de vasculite (mais claramente demonstrada na doença associada a ANCA), prescreve-se metotrexato (com folato) ou azatioprina diária para substituir a ciclofosfamida porque esses fármacos têm um perfil de efeitos colaterais melhor. Também pode-se utilizar rituximabe IV periodicamente para manter a remissão, mas a dose ideal e o intervalo de infusão não foram claramente estabelecidos. A duração do tratamento varia de um a vários anos, dependendo do paciente, diagnóstico específico e propensão a recidivas. Pacientes com recidivas frequentes podem precisar tomar imunossupressores indefinidamente.
O uso prolongado de corticoides pode ter efeitos colaterais significativos. Pacientes tomando ≥ 7,5 mg de prednisona por dia ou doses equivalentes de outros corticoides devem receber suplementos de cálcio, vitamina D e bisfosfonatos para ajudar a prevenir ou minimizar a osteoporose; deve-se considerar o monitoramento da densidade óssea. Pode ser necessária suplementação de corticoides adicionais em pacientes gravemente enfermos ou naqueles submetidos à cirurgia que podem ter um eixo hipotálamo-hipófise-adrenal suprimido, dependendo da dose e duração da terapia com corticoides e duração e intensidade do estresse.
Estudos recentes voltaram-se ao desenvolvimento de tratamentos que limitam a exposição a corticoides. Avacopan, um antagonista seletivo do receptor C5a, é um tratamento adjuvante disponível para a vasculite ativa grave associada a ANCA (1).
Referência sobre o tratamento
1. Jayne DRW, Merkel PA, Schall TJ, et al: Avacopan for the treatment of ANCA-associated vasculitis. N Engl J Med 384(7):599-609, 2021. doi: 10.1056/NEJMoa2023386. PMID: 33596356
Pontos-chave
A vasculite pode ser uma doença primária ou secundária a outras causas.
A vasculite tende a atingir vasos pequenos, médios ou grandes, cada um com certos padrões de comprometimento de órgãos.
As manifestações clínicas podem ser sistêmicas e/ou específicas de cada órgão, dependendo do comprometimento dos vasos.
Fazer exames de sangue, exames de imagem e biópsia de tecido segundo a indicação a fim de determinar a causa da vasculite (incluindo as infecções e as neoplasias), a extensão do comprometimento dos órgãos e a gravidade da doença.
Tratar com corticoides e imunossupressores.
Abordar aumento do risco de infecção e de osteoporose causada pelo tratamento da vasculite com monitoramento e/ou profilaxias.







