




Os tumores ósseos malignos primários são muito menos comuns que os tumores metastáticos, principalmente em adultos. Tumores ósseos malignos primários incluem mieloma múltiplo, osteossarcoma, adamantinoma, condrossarcoma, cordoma, Sarcoma de Ewing do osso, fibrossarcoma e sarcoma pleomórfico indiferenciado, linfoma do osso e tumor maligno de células gigantes. (Ver também Visão geral dos tumores ósseos e articulares e Visão geral da leucemia.)
Os dois sistemas mais amplamente utilizados para o estadiamento desses tumores são
The American Joint Committee on Cancer (AJCC) Cancer Staging Manual, 8ª edição: para osteosarcoma, condrossarcoma e sarcoma de Ewing, o estadiamento baseia-se em categorias tumorais distintas, grau histológico, tamanho, envolvimento nodal e metástases (classificação TNM). O manual classifica os tumores em 4 estádios e é utilizado para documentar dados sobre câncer.
O sistema de estadiamento Musculoskeletal Tumor Society (MSTS): utilizado por cirurgiões oncológicos ortopédicos com base no grau histológico (p. ex., histologia de estádio I de baixo grau e histologia de estádio II de alto grau, se o tumor está totalmente contido no osso (A) ou se rompeu fora do córtex para partes moles adjacentes (B) e metástases, estádio III). O osteossarcoma típico (convencional) com uma massa de tecidos moles associada sem metástases é o estádio IIB no sistema MSTS.
Mieloma múltiplo
O mieloma múltiplo é o tumor ósseo maligno primário mais comum, mas frequentemente é considerado um tumor das células da medula óssea em vez de um tumor primário ósseo por ter origem hematopoiética (Ver também Mieloma múltiplo). Mesmo que o mieloma múltiplo seja considerado um tumor hematológico, deve-se diferenciar a anormalidade esquelética identificada de outros tumores ósseos.
Mieloma múltiplo ocorre principalmente em idosos.
O desenvolvimento e o progresso tumoral costumam ser multicêntricos e, com frequência, envolvem a medula óssea tão difusamente que o diagnóstico é feito por punção. Contrariamente ao que ocorre na doença metastática, a cintilografia óssea com radionuclídeo pode não mostrar as lesões com fidedignidade e deve-se outros fazer exames do osso. Os exames do osso normalmente mostram lesões líticas circunscritas bem definidas (lesões em saca-bocado) ou desmineralização difusa. Raramente a lesão pode aparecer como esclerótica ou como osteopenia difusa, especialmente em um corpo vertebral. Uma lesão isolada de mieloma único sem envolvimento sistêmico da medula é denominada plasmocitoma.

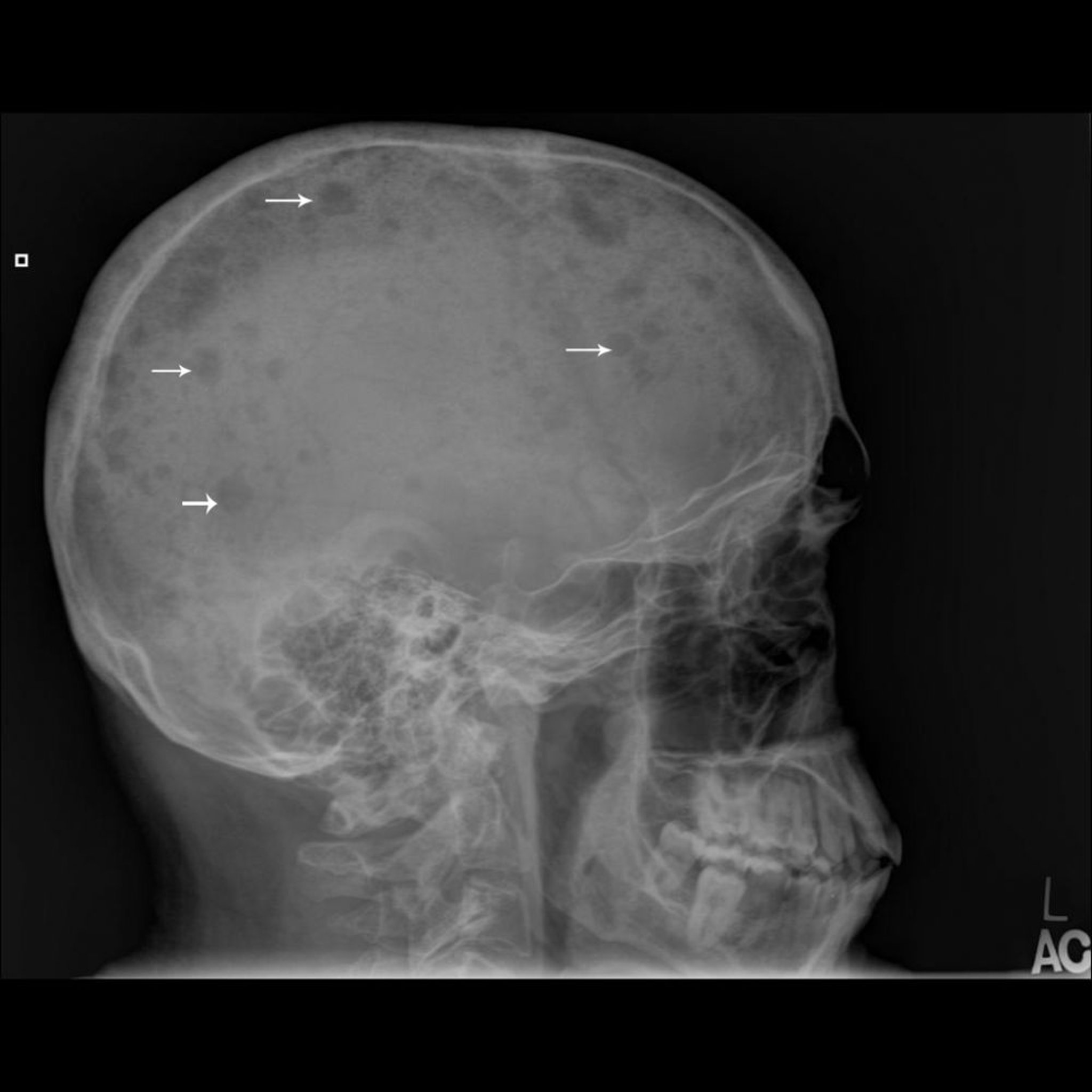
Imagem cedida por cortesia de Michael J. Joyce, MD, e Hakan Ilaslan, MD.
Certas lesões ósseas respondem muito bem à radioterapia.
Osteossarcoma (sarcoma osteogênico)
Osteossarcoma é o tumor ósseo maligno primário mais comum (se considerarmos o mieloma um tumor de células da medula e não um tumor ósseo primário) e é altamente maligno. É mais comum em pessoas de 10 a 25 anos de idade, embora possa ocorrer em qualquer idade. Há dois picos de incidência; a incidência é mais alta em adolescentes e adultos muito jovens (coincidindo com o estirão de crescimento da adolescência) e o pico secundário ocorre em idosos (≥ 60 anos), especialmente naqueles com fatores de risco como doença de Paget, infartos ósseos e áreas do osso previamente expostas a altas doses de radioterapia para outro câncer muitos anos antes. Há uma predisposição genética, sobretudo em crianças portadoras do gene para retinoblastoma hereditário (variantes do gene RB1) e síndrome de Li-Fraumeni (gene TP53).
O osteossarcoma produz osteoide maligno (osso imaturo) a partir de células ósseas com tumor. O osteossarcoma normalmente se desenvolve ao redor do joelho (fêmur distal mais frequente do que tíbia proximal) ou em outros ossos longos, particularmente nas metáfises e diáfises e pode criar metástase em pulmão ou osso. Dor e edema são sintomas comuns.

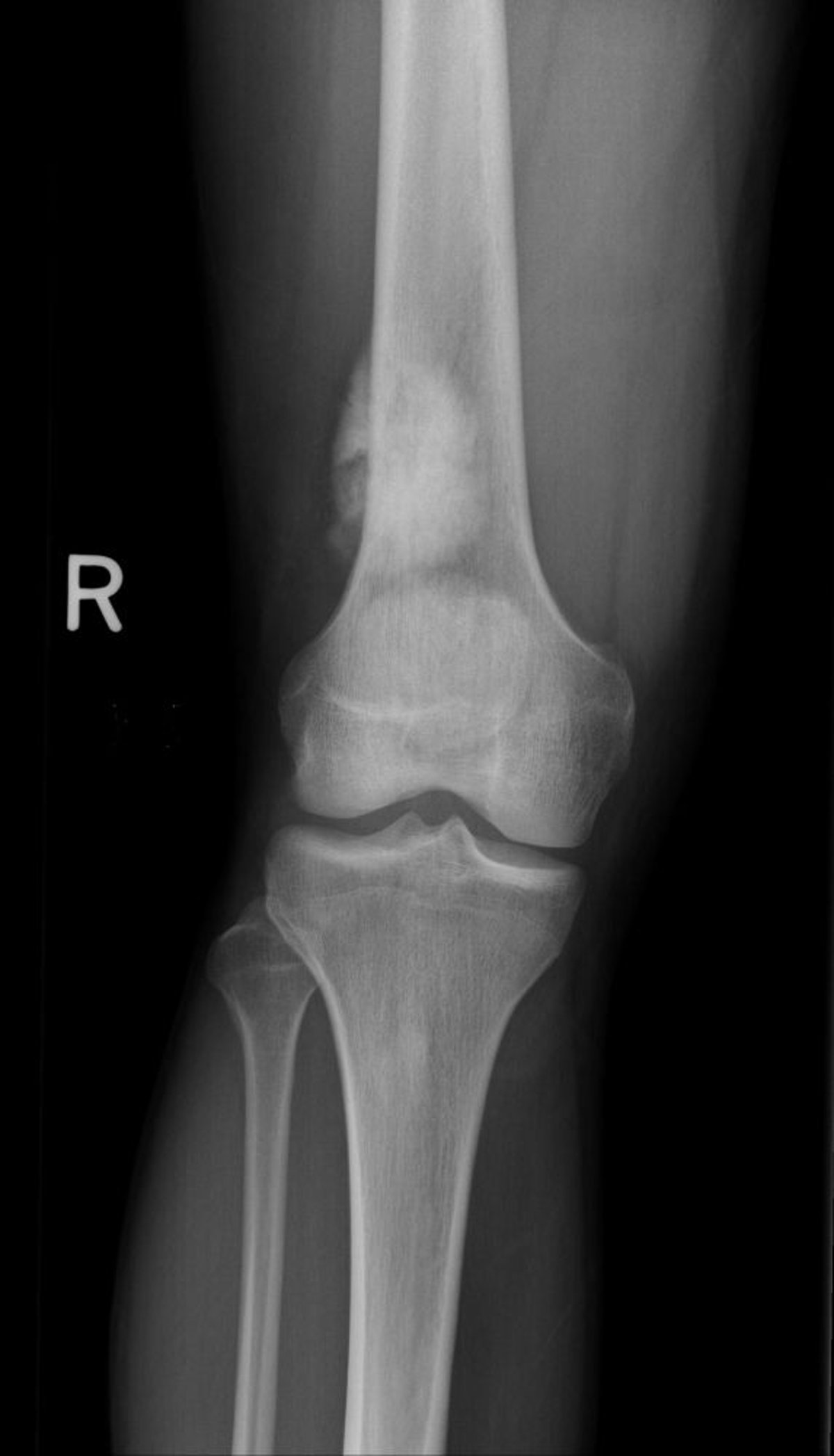
Image courtesy of Michael J. Joyce, MD, e Hakan Ilaslan, MD.
Os achados radiográficos variam e podem incluir características escleróticas ou líticas. O diagnóstico do osteossarcoma exige a biópsia. Os pacientes necessitam de radiografia torácica e TC para detectar metástases pulmonares, além de exame ósseo para detectar metástase óssea. Fazer a RM de todo o membro comprometido a fim de avaliar se há lesões metacrônicas. PET-TC pode mostrar metástases ou lesões metacrônicas a distância.
O tratamento do osteossarcoma é feito pela combinação de quimioterapia e cirurgia. O tratamento com quimioterapia adjuvante aumenta a sobrevida de < 20% a > 65% em 5 anos. A quimioterapia neoadjuvante é iniciada antes de qualquer resseção cirúrgica. Diminuição da massa tumoral periférica de tecidos moles ou aumento da mineralização nas radiografias, redução do nível de dor e do nível sérico de fosfatase alcalina indicam alguma resposta, mas a resposta desejada é > 95% de necrose tumoral no mapeamento histológico da amostra resseccionada coletada pelo patologista. Após várias sessões de quimioterapia (vários meses), a cirurgia com preservação do membro pode proceder. Ocasionalmente, realiza-se amputação cirúrgica antes do início da quimioterapia para tumor fúngico. O objetivo é tratar precocemente a doença micrometastática que supostamente está presente, mesmo que ela não seja visível nos exames de imagem de estadiamento.
Nessa cirurgia com preservação do membro, o tumor é resseccionado em bloco, incluindo todo o tecido reativo ao redor e uma borda do tecido normal circundante; para evitar extravasamento de células tumorais, o tumor não é violado. Mais de 85% dos pacientes podem ser tratados com cirurgia de reconstrução sem diminuição da sobrevida em longo prazo.
A continuação da quimioterapia é necessária. Se a necrose tumoral estiver quase completa (cerca de 95%) devido a quimioterapia pré-operatória, sobrevida é de > 90% em 5 anos. A doença metastática limitada aos pulmões às vezes pode ser tratada com toracotomia e resseção em cunha da lesão pulmonar.
As variantes do osteossarcoma que diferem do osteossarcoma convencional e ocorrem com muito menos frequência incluem lesões da superfície cortical, como o osteossarcoma parosteal e o osteossarcoma periosteal. Osteossarcoma parosteal na maioria das vezes compromete o córtex posterior da região distal do fêmur sendo geralmenterelativamente bem diferenciados. Quimioterapia não é necessária antes da ressecção cirúrgica para o tratamento de osteossarcoma parosteal de baixo grau. Osteossarcomas parosteais exigem ressecção cirúrgica em bloco, mas não quimioterapia se a histologia da amostra ressecada confirmar que o tumor é bem diferenciado.
Osteossarcoma periosteal é mais um tumor de superfície da matriz da cartilagem que também contém matriz óssea e é maligno. Muitas vezes se localiza no eixo central do fêmur e aparece como um rajada de luz nas radiografias. A probabilidade de metástases dos osteossarcomas periosteais é muito maior do que a dos osteossarcomas parosteais bem diferenciados, mas um pouco menor do que a dos osteossarcomas típicos. Na maioria das vezes, os osteossarcomas periosteais são tratados de modo semelhante aos osteossarcomas convencionais com quimioterapia e ressecção cirúrgica em bloco.
Adamantinoma
O adamantinoma é raro (< 1% dos tumores ósseos malignos) e, geralmente, se desenvolve na tíbia. Normalmente ocorre em adolescentes e em pessoas entre os 20 e 30 anos, mas pode ocorrer em qualquer idade. O adamantinoma é de crescimento lento e costuma se manifestar com dor e plenitude palpável.

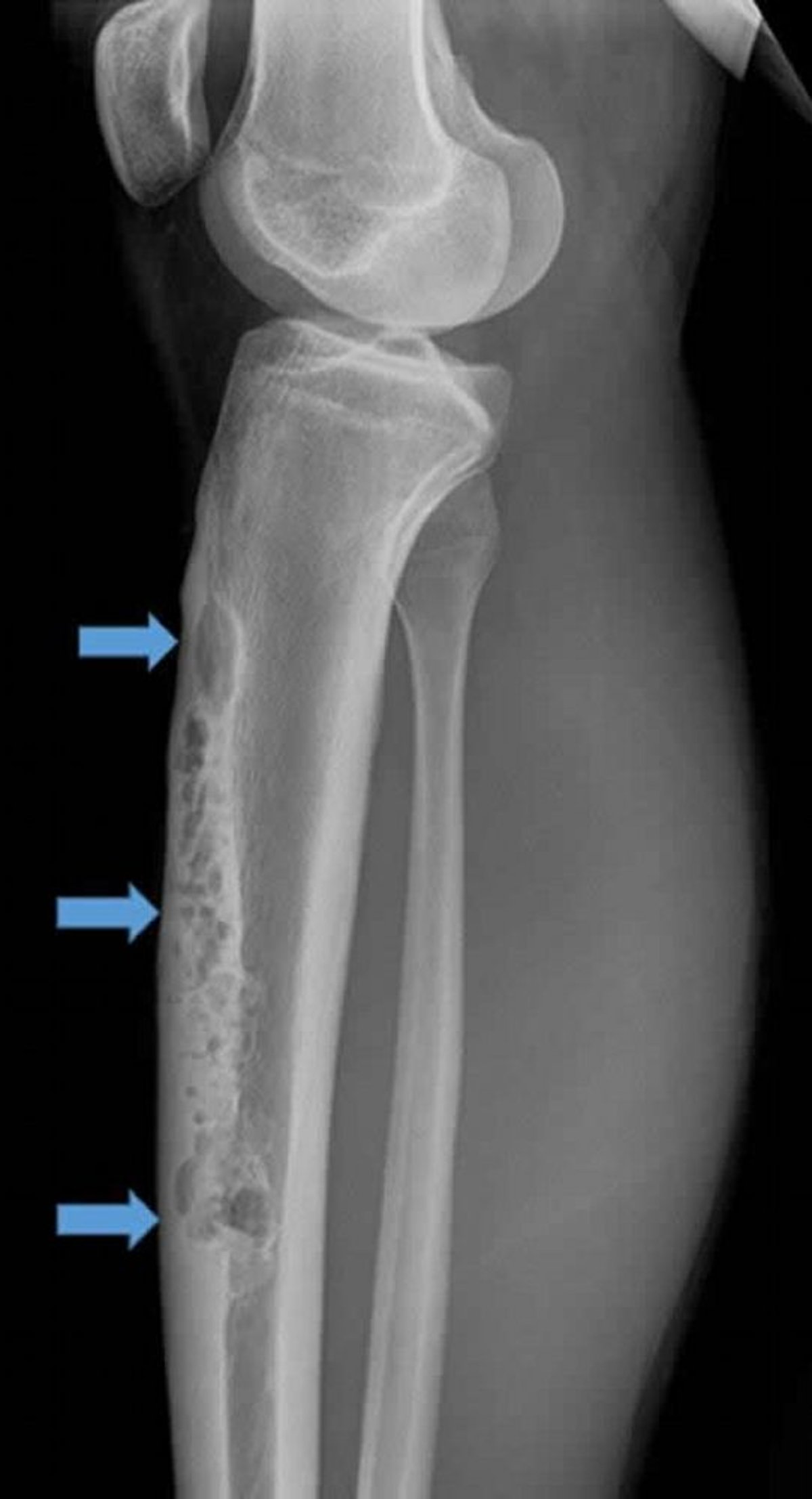
Image courtesy of Michael J. Joyce, MD, e Hakan Ilaslan, MD.
A lesão geralmente se manifesta na crista anterior da tíbia, e radiografias mostram uma aparência osteolítica de "bolha de sabão". A aparência histológica é um padrão bifásico de tecido epitelial e osteofibroso. A lesão pode ser confundida com displasia osteofibrosa do córtex tibial anterior, que é benigno. Alguns médicos acham que a displasia osteofibrosa do córtex tibial anterior pode ser um precursor do adamantinoma, mas sem o componente epitelial que o tornaria um câncer.
Ocorrem metástases, principalmente nos pulmões, mas são raras.
O tratamento da adamantinoma consiste em excisão ampla e reconstrução do defeito. Às vezes, é necessária amputação.
Condrossarcoma
Os condrossarcomas são tumores malignos da cartilagem. Eles diferem clínica, terapêutica e prognosticamente do osteossarcoma. Nos condrossarcomas, 90% são tumores primários. Os condrossarcomas também podem surgir em outras doenças preexistentes, particularmente nos osteocondromas múltiplos (p. ex., na doença de Ollier e na síndrome de Maffucci). Os condrossarcomas tendem a ocorrer em idosos. Eles geralmente se desenvolvem em ossos chatos (p. ex., pelve, escápula), mas podem se desenvolver em qualquer parte de qualquer osso (mais frequentemente o fêmur e o úmero entre os ossos longos) e podem ter um componente de tumor de tecidos moles envolvendo os tecidos moles circundantes.

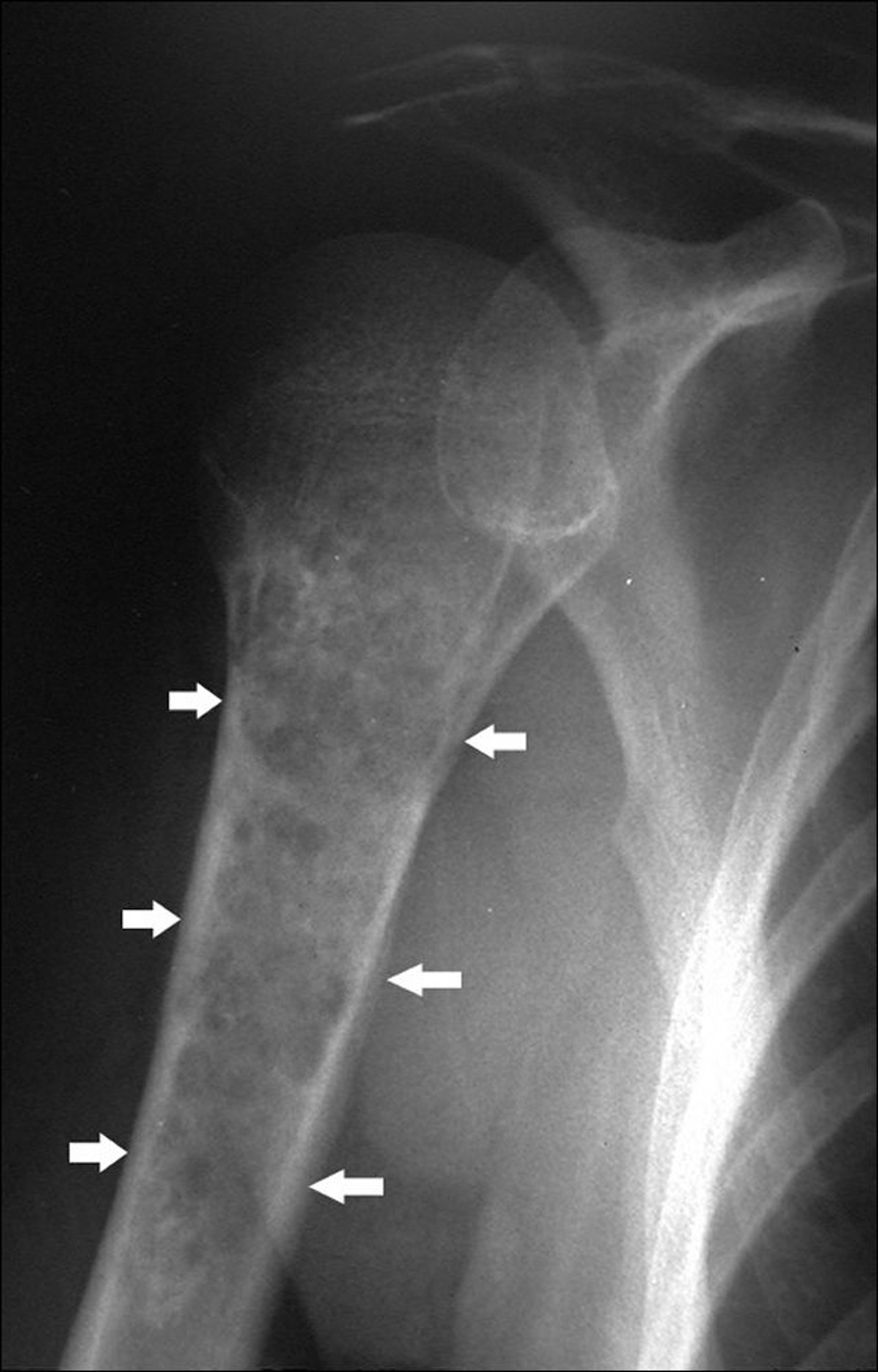
Image courtesy of Michael J. Joyce, MD, e Hakan Ilaslan, MD.
Radiografias simples normalmente revelam pontos de calcificações. Os condrossarcomas também exibem, com frequência, destruição da cortical óssea e perda das trabéculas ósseas normais. A RM pode mostrar massa em tecidos moles. Cintilografia também pode ser feita. O condrossarcoma exige diagnóstico tecidual, que também pode determinar o estádio do tumor (probabilidade de metástases). A biópsia por agulha pode oferecer uma amostra inadequada de tecido.
Muitas vezes é difícil diferenciar os condrossarcomas de baixo grau dos enchondromas pelos exames de imagem, e algumas vezes até mesmo pela citopatologia.
Os condrossarcomas de baixo grau (grau 1) são frequentemente tratados de forma intralesional (ampla curetagem) com acréscimo de um adjuvante (frequentemente nitrogênio líquido congelante, laser de argônio, calor de metilmetacrilato, radiofrequência ou fenol). Alguns cirurgiões preferem ressecção cirúrgica em bloco dos tumores de baixo grau a fim de reduzir o risco de recidiva. Os tumores de grau mais alto são tratados por meio da ressecção cirúrgica em bloco. Quando a ressecção cirúrgica com manutenção da função é impossível, a amputação pode ser necessária. Devido ao potencial para implante, deve-se ter cuidado para evitar a disseminação das células do tumor nos tecidos moles quando feita a biópsia ou a cirurgia. A recorrência é inevitável se as células do tumor se disseminarem. Se elas não se disseminarem, a chance de cura depende do grau do tumor. Tumores com graus baixos são quase todos curados com o devido tratamento. Como esses tumores tem vascularidade limitada, a quimioterapia e radioterapia tem pouca eficácia.
Cordoma
O cordoma, que é raro, se desenvolve de restos de notocorda primitiva. Isso tende a ocorrer nas extremidades da coluna espinal, normalmente no sacro ou perto da base do crânio. O cordoma na região sacrococcígea produz dor quase constante. O cordoma na base do crânio pode produzir deficits em qualquer nervo cranial, mais comum nos nervos dos olhos.
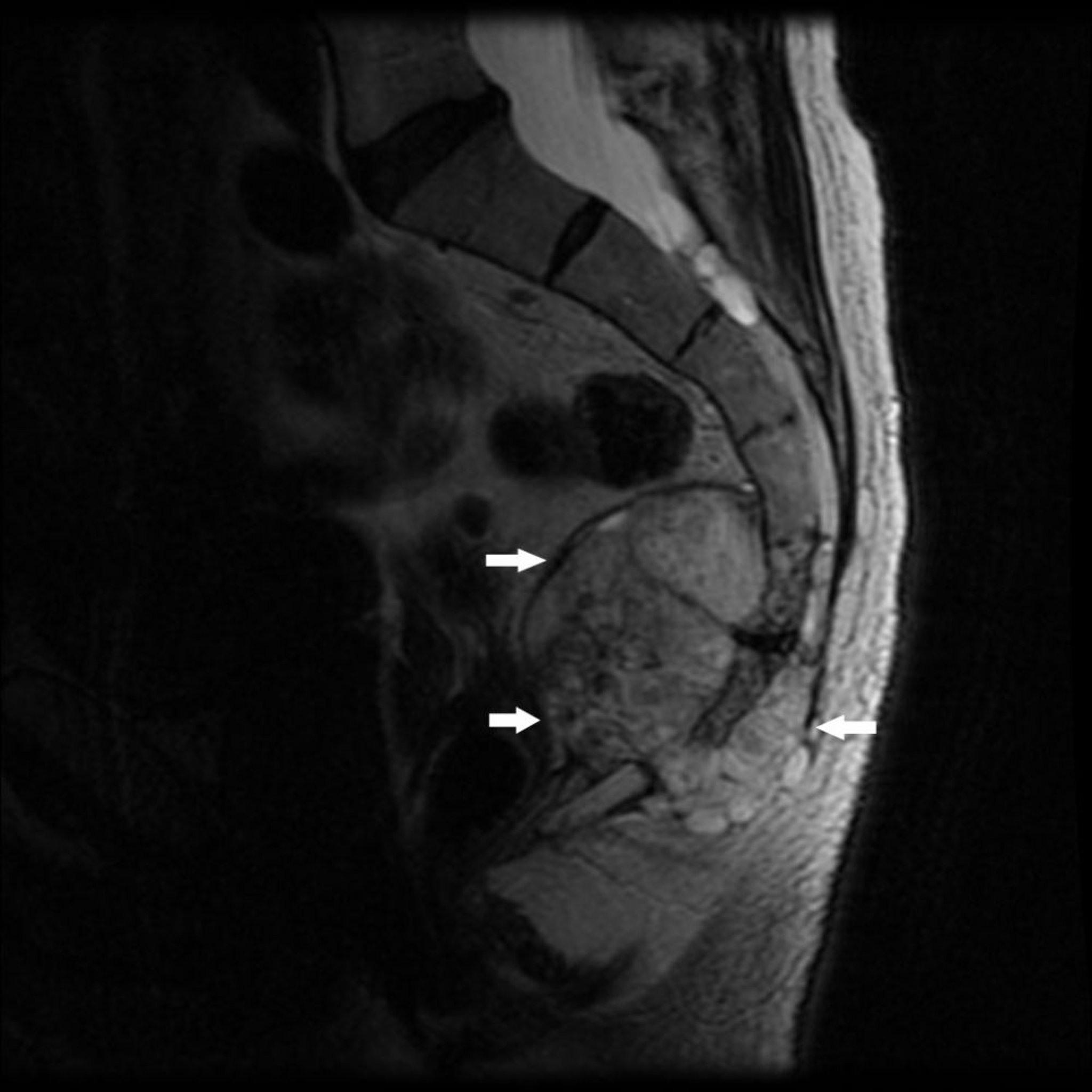
Image courtesy of Michael J. Joyce, MD, e Hakan Ilaslan, MD.
Pode haver sinais e sintomas do cordoma meses a muitos anos antes do diagnóstico.
O cordoma aparece nas radiografias como lesão óssea destrutiva que pode estar associada a massa de tecidos moles. Biópsia é feita.
Os cordomas na região sacrococcígea podem ser curados pela excisão radical total. Os cordomas na base do crânio são normalmente inacessíveis à cirurgia, mas podem responder à radioterapia. A taxa de recorrência local é alta após a ressecção tumoral. Metástases, embora menos comuns, podem ocorrer.
Tumor de Ewing do osso
O sarcoma ósseo de Ewing é um pequeno tumor ósseo de células redondas de cor azulada, com pico de incidência entre 10 e 20 anos. O sarcoma de Ewing está relacionado com tumores neuroectodérmicos primitivos periféricos (TNEP) e com o tumor maligno de células pequenas de Askin da parede torácica, que agora são considerados parte da família dos sarcomas de Ewing. A maioria dos tumores se desenvolve nas extremidades, mas qualquer osso pode ser envolvido. O tumor de Ewing tende a ser extenso, algumas vezes comprometendo todo o eixo do osso, mais comumente, a diáfise. Cerca de 15 a 20% ocorre ao redor da metáfise. Dor e edema são os sintomas mais comuns.

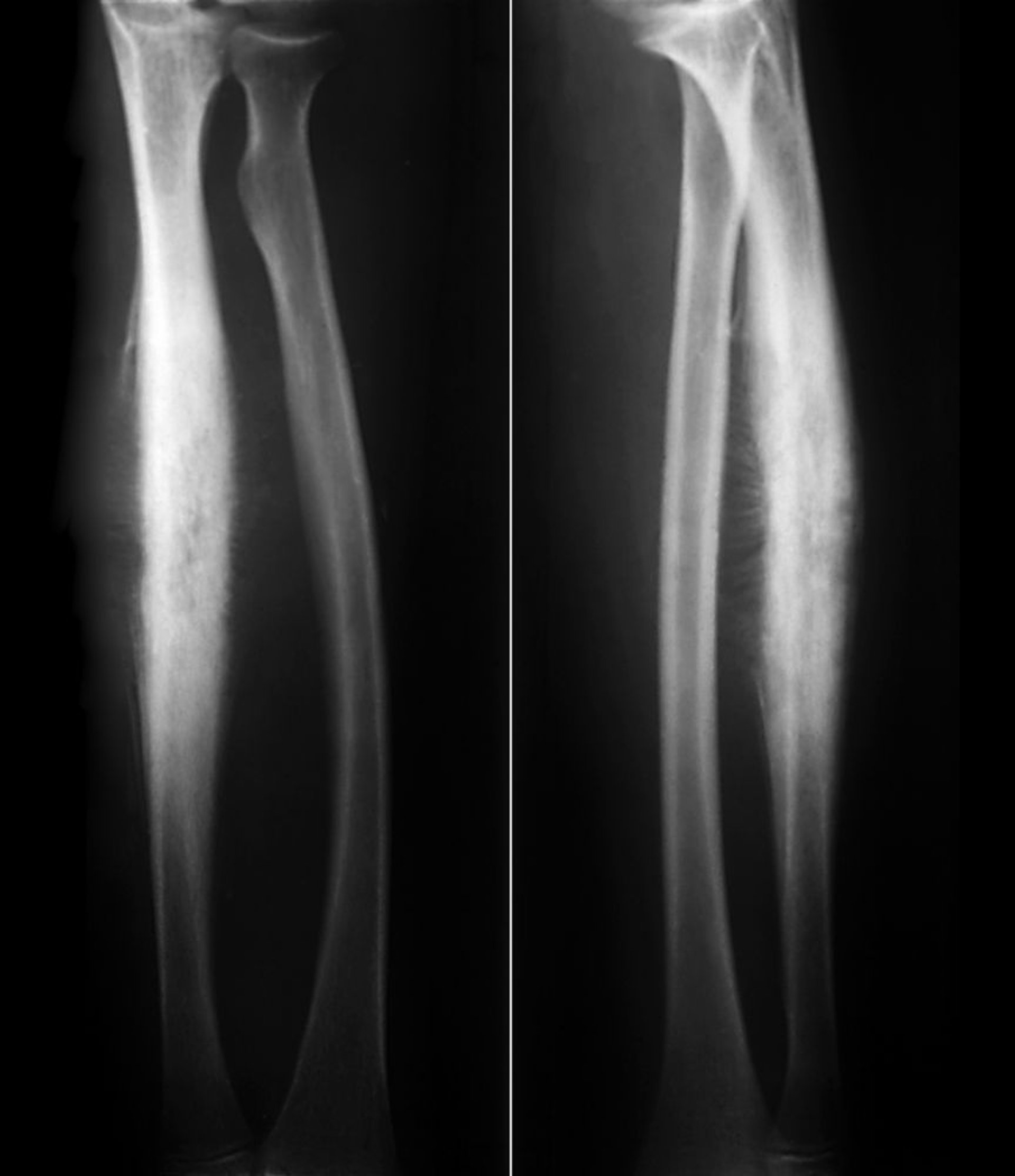
ZEPHYR/SCIENCE PHOTO LIBRARY
As destruições líticas, particularmente um padrão de infiltração permeativa sem margens definidas consistem no achado mais comum em radiografias, mas as camadas múltiplas da formação óssea nova de subperiósteo reativo podem dar aparência de “pele-de-cebola”. Normalmente, os radiografias simples não revelam a inteira extensão envolvida e uma grande massa de tecido mole geralmente circundam o osso afetado. RM define melhor a extensão da doença, podendo ajudar a guiar o tratamento.
Muitos outros tumores benignos e malignos podem parecer semelhantes, de modo que o diagnóstico do sarcoma de Ewing deve ser feito por biópsia. Às vezes, esse tipo de tumor pode ser confundido com uma infecção. Pode-se alcançar um diagnóstico histológico preciso com análise citogenética e identificação de marcadores moleculares, incluindo avaliação para anormalidade cromossômica clonal típica. Os achados moleculares na família de tumores de Ewing são translocações cromossômicas não aleatórias distintas envolvendo o gene do sarcoma de Ewing (SEW) no cromossomo 22. Identificaram-se 18 diferentes translocações estruturais em diferentes padrões de genes de fusão.

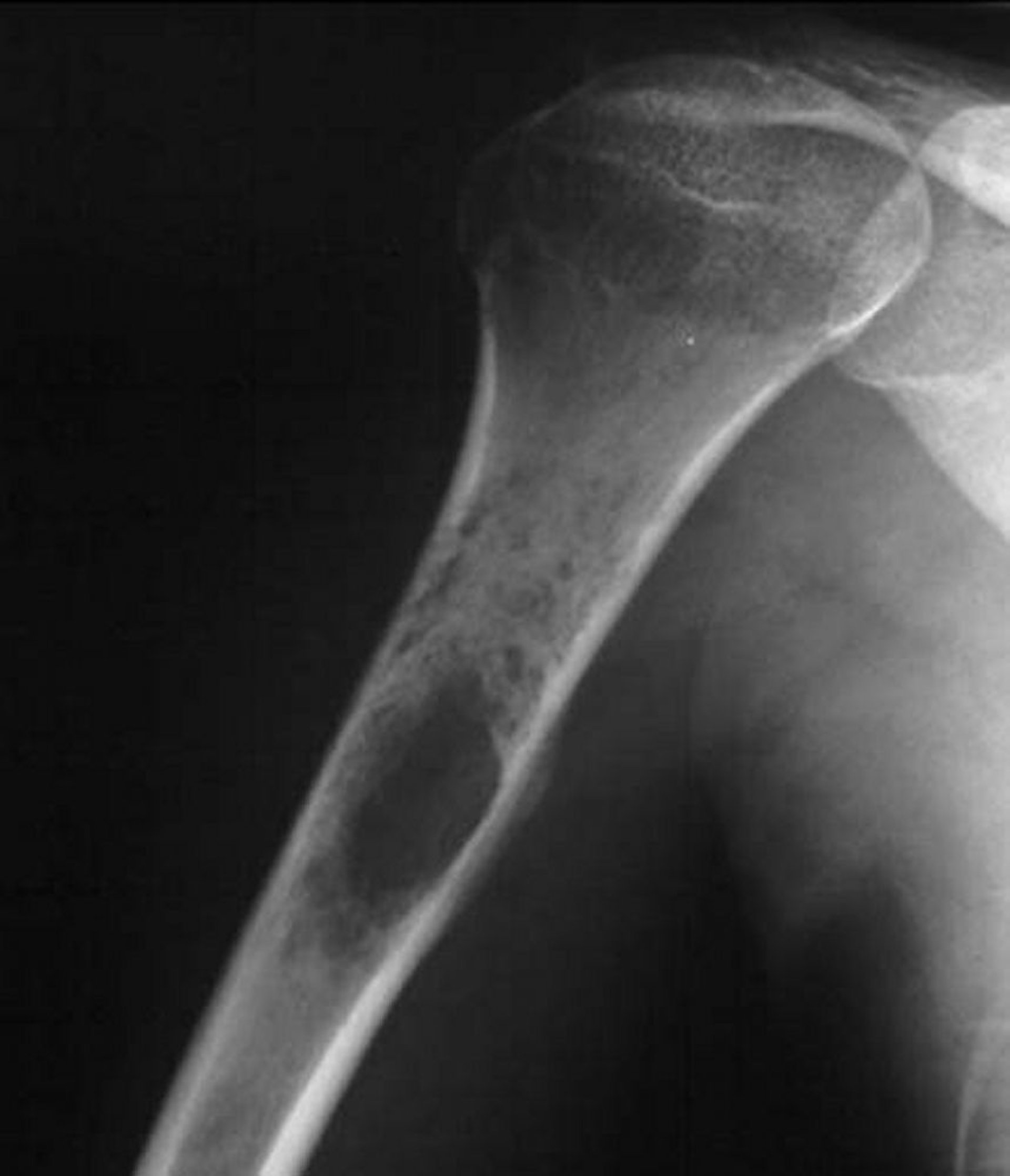
Image courtesy of Michael J. Joyce, MD, e Hakan Ilaslan, MD.
O tratamento do sarcoma de Ewing é feito com várias combinações de cirurgia, quimioterapia e radioterapia. Atualmente, > 60% dos pacientes com tumor de Ewing primário localizado podem ser curados por essa abordagem multimodal. A cura é possível de vez em quando, mesmo que a doença seja metastática. Quimioterapia em conjunto com ressecção cirúrgica em bloco, se aplicável, geralmente produz melhores resultados a longo prazo do que a quimioterapia em conjunto com radioterapia.
Fibrossarcoma e sarcoma pleiomórfico indiferenciado (antigo histiocitoma fibroso maligno do osso)
Os fibrossarcomas e o sarcoma pleiomórfico indiferenciado têm características semelhantes aos osteossarcomas, mas produzem células tumorais fibrosas (em vez de células tumorais ósseas), ocorrem na mesma faixa etária e apresentam problemas similares.
O tratamento e o desfecho das lesões de alto grau são semelhantes aos dos osteossarcomas.
Linfoma ósseo
O linfoma ósseo (previamente conhecido como sarcoma da célula do retículo) afeta adultos, normalmente entre 40 e 60 anos de idade. Isso costuma ser um linfoma difuso de grandes células B. Pode crescer em qualquer osso. O tumor consiste em pequenas células redondas, normalmente com mistura das células do retículo, linfoblastos e linfócitos. Pode se desenvolver em tumor ósseo primário isolado, em associação a tumores similares em outros tecidos ou como metástase de doença conhecida de tecido linfomatoso mole. Dor e edema são os sintomas habituais do linfoma ósseo. A fratura patológica é comum.

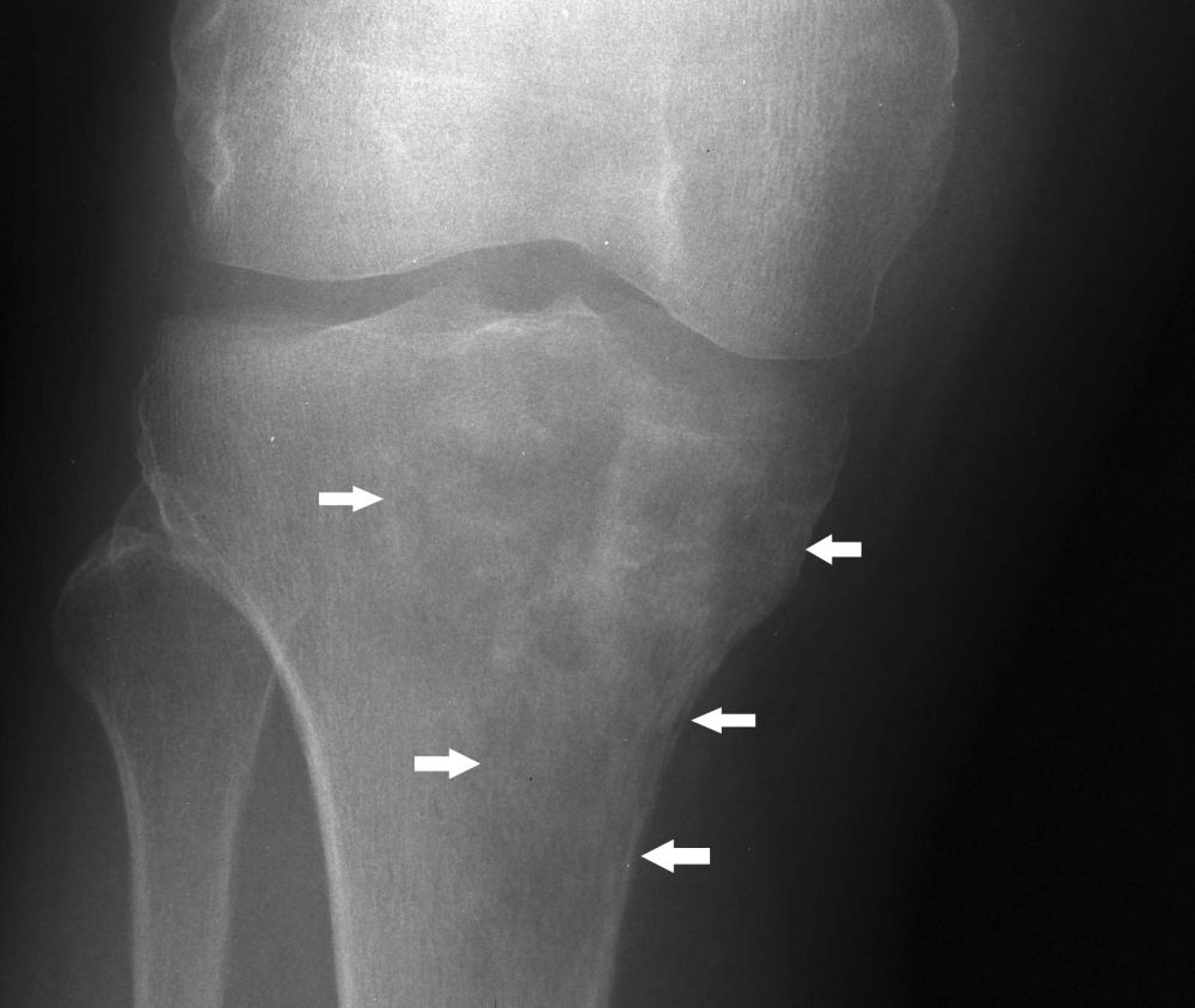
Image courtesy of Michael J. Joyce, MD, e Hakan Ilaslan, MD.
As radiografias revelam destruição óssea, a qual podes apresentar padrão permeável manchado ou irregular, ou mesmo infiltrado, frequentemente com grande massa de tecido mole radiográfica e clínica. Na doença avançada, o contorno inteiro do osso afetado pode estar perdido. Biópsia também é feita.
No linfoma ósseo primário isolado, a sobrevida em 5 anos é ≥ 50%.
Os linfomas ósseos são tipicamente tratados com quimioterapia sistêmica. Em alguns casos há indicação de radioterapia adjuvante. A estabilização de ossos longos é frequentemente necessária para prevenir fratura patológica. A amputação é indicada apenas raramente, quando a função é perdida devido à fratura patológica ou ao envolvimento extensivo do tecido mole, os quais não podem ser monitorados de outra maneira.
Tumor maligno de célula gigante
O tumor maligno de célula gigante, que é raro, é normalmente localizado no final extremo do osso longo.
Radiografias revelam características clássicas de destruição maligna (predominantemente destruição lítica, destruição cortical, extensão do tecido mole e fratura patológica). O tumor maligno de célula gigante que se desenvolve previamente em um tumor benigno da célula gigante é caracteristicamente radiorresistente. RM e biópsia são realizadas.
O tratamento do tumor maligno de células gigantes é semelhante ao do osteossarcoma, mas os índices de cura são baixos.







