




La neoplasia endocrina múltiple tipo 2A (NEM 2A) es un síndrome autosómico dominante caracterizado por el hallazgo de carcinoma medular de tiroides, feocromocitoma, hiperplasia o adenomas paratiroideos (que causan hiperparatiroidismo) y, en ocasiones, amiloidosis liqueniforme cutánea. Las características clínicas dependen de los elementos glandulares comprometidos. El carcinoma familiar medular de la glándula tiroides es una variedad de la NEM 2A. El diagnóstico se confirma con los estudios genéticos. Las pruebas hormonales y de diagnóstico por la imagen contribuyen a la localización del tumor, que debe extirparse quirúrgicamente siempre que sea posible.
(Véase también Generalidades sobre las neoplasias endocrinas múltiples.)
Se identificaron mutaciones en el protooncogén RET en el cromosoma 10 en pacientes con NEM 2A, NEM 2B y carcinoma familiar medular de la glándula tiroides. La proteína RET es una tirosina cinasa receptora; las mutaciones asociadas con la NEM 2A y el carcinoma familiar medular de la glándula tiroides promueven la activación de ciertas vías intracelulares específicas.
Síntomas y signos de la NEM 2A
Las características clínicas dependen del tipo tumoral (véase tabla Condiciones asociadas con los síndromes de neoplasia endocrina múltiple).
Tiroides
Casi todos los pacientes presentan un carcinoma medular de la glándula tiroides. El tumor suele aparecer durante la infancia y comienza con una hiperplasia de las células C parafoliculares de la tiroides. A menudo, los tumores son multicéntricos.
Glándulas suprarrenales
El feocromocitoma suele originarse en las glándulas suprarrenales. En el 40 al 50% de las personas emparentadas con NEM 2A se identifica feocromocitoma. A diferencia del feocromocitoma esporádico, la variedad familiar dentro de la NEM 2A comienza con una hiperplasia de la médula suprarrenal y es multicéntrica y bilateral en > 50% de los casos. Los feocromocitomas son infrecuentes fuera de las glándulas suprarrenales. Estos tumores casi siempre son benignos, pero tienden a recidivar en forma local y producen una morbimortalidad significativa.
Los feocromocitomas que se desarrollan en pacientes con NEM 2A (y 2B) suelen producir adrenalina en concentraciones desproporcionadas respecto de la noradrenalina, lo que los diferencia de los casos esporádicos.
Las crisis hipertensivas provocadas por el feocromocitoma son un manifestación habitual. La hipertensión identificada en pacientes con NEM 2A y feocromocitoma suele presentarse en forma paroxística con mayor frecuencia que sostenida, a diferencia de lo que se observa en los casos esporádicos habituales. Los pacientes con feocromocitomas pueden experimentar episodios paroxísticos caracterizados por palpitaciones, ansiedad, cefaleas o sudoración, aunque muchos son asintomáticos.
Paratiroides
Entre el 10 y el 20% de los pacientes presenta evidencias de hiperparatiroidismo (que puede ser de larga data) con hipercalcemia, nefrolitiasis, nefrocalcinosis o insuficiencia renal. El hiperparatiroidismo compromete con frecuencia varias glándulas en forma de hiperplasia generalizada o de múltiples adenomas y, en los pacientes con NEM 2A, pueden detectarse trastornos leves de la función paratiroidea.
Otras manifestaciones
En algunas familias con NEM 2A, puede identificarse amiloidosis liqueniforme cutánea, que se caracteriza por una lesión cutánea papular pruriginosa y escamosa localizada en la región interescapular o sobre las superficies extensoras. La enfermedad de Hirschsprung está presente en 2 a 5% de los pacientes con NEM 2A.

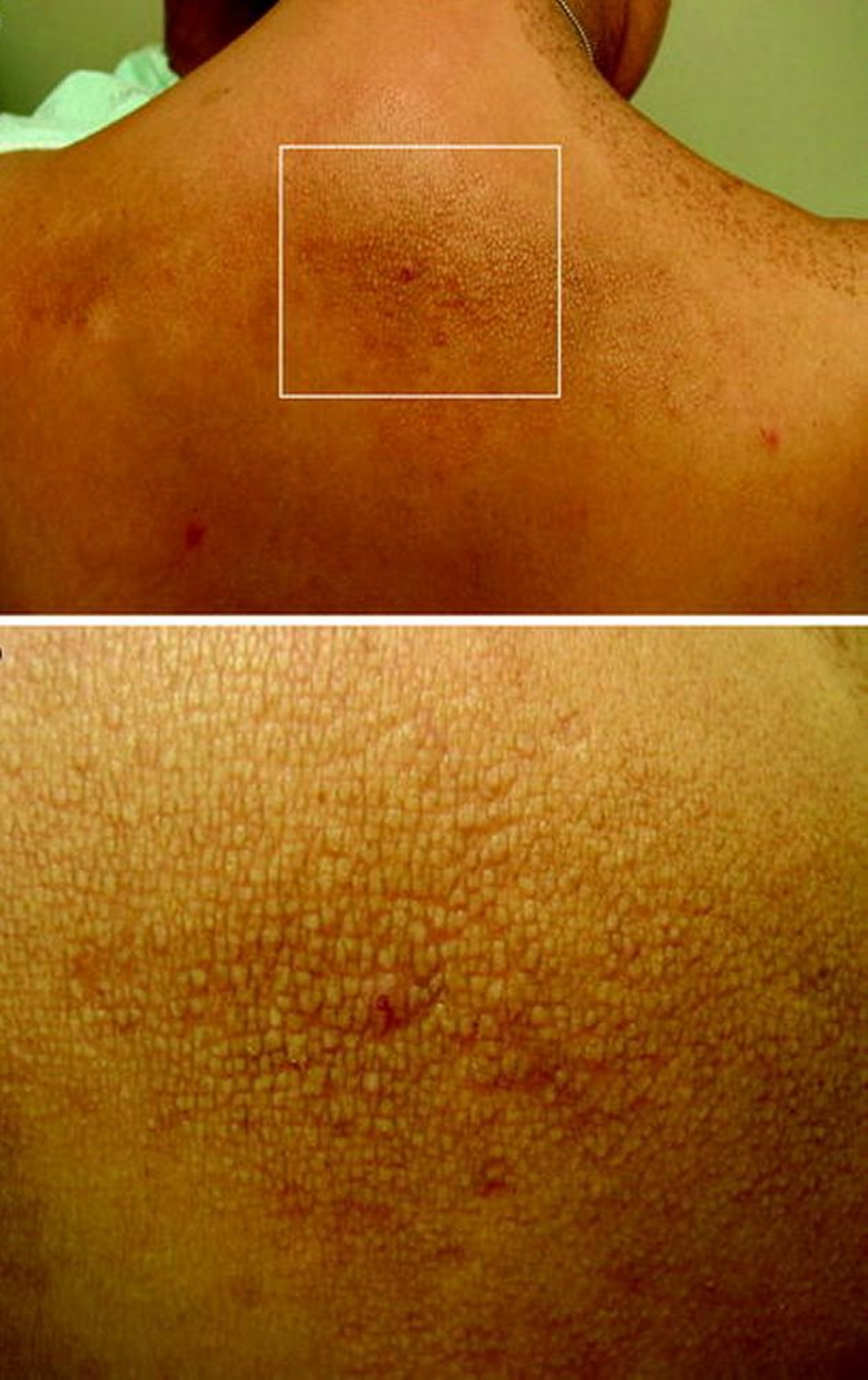
© Springer Science+Business Media

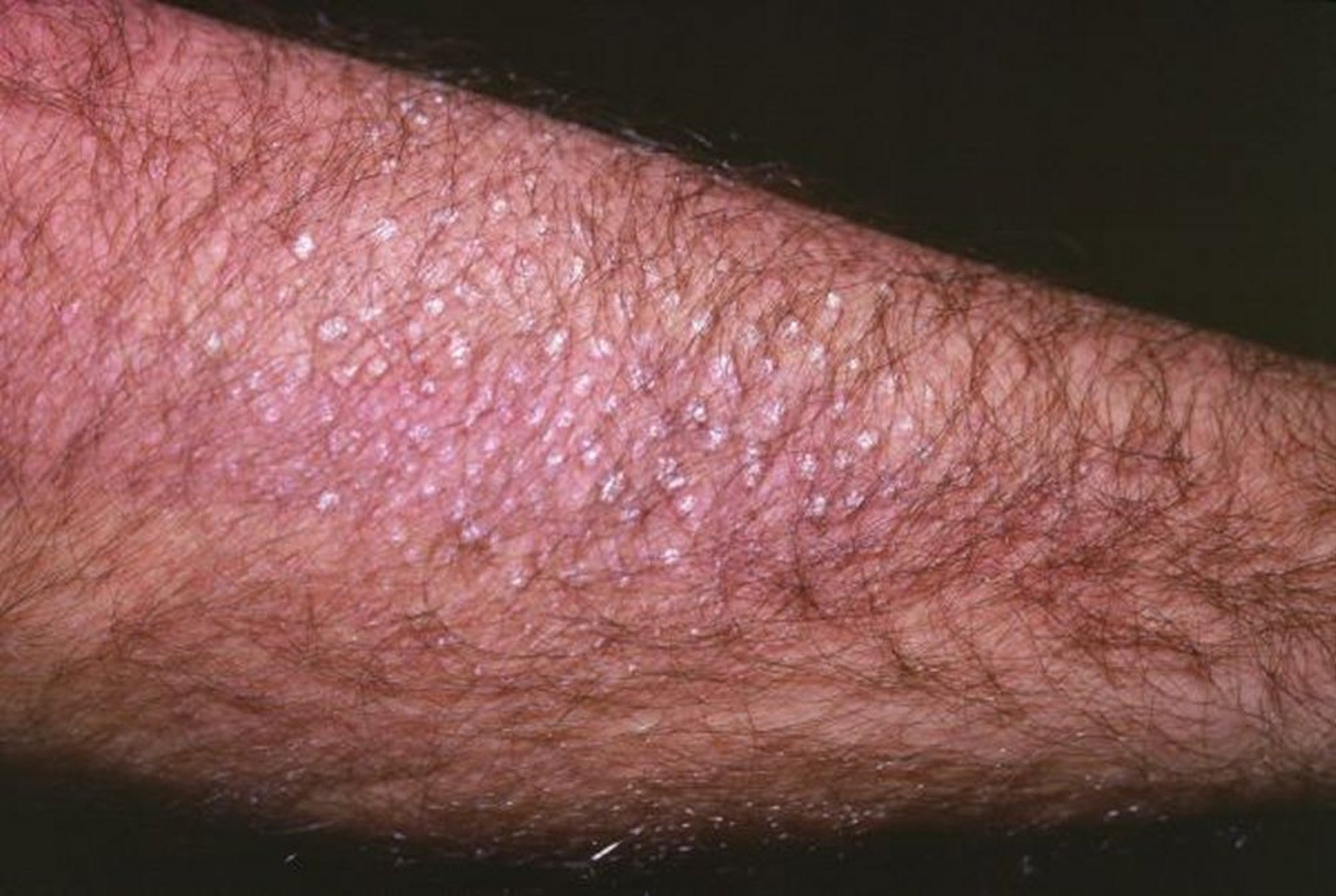
Imagen cortesía de Karen McKoy, MD.
Diagnóstico de la NEM 2A
Calcitonina sérica para el carcinoma medular de tiroides
Calcio sérico, calcio en orina de 24 horas y hormona paratiroidea para el hiperparatiroidismo
Metanefrinas libres en plasma o niveles urinarios de catecolaminas y metanefrinas para detectar un feocromocitoma
Ecografía del cuello, con imágenes adicionales según sea necesario
Localización del feocromocitoma con RM o TC
Estudios genéticos
La calcitonina sérica es un buen marcador de la presencia de carcinoma medular de tiroides y también puede usarse como biomarcador para seguir la respuesta y/o la progresión de la enfermedad.
El hiperparatiroidismo se diagnostica ante el hallazgo de hipercalcemia, hipofosfatemia y aumento de la concentración de hormona paratiroidea.
Dado que el feocromocitoma puede ser asintomático, su exclusión puede ser difícil. Las pruebas más sensibles para el diagnóstico son la medición de las concentraciones plasmáticas de metanefrinas libres o las concentraciones urinarias fraccionadas de catecolaminas y metanefrinas (en particular, adrenalina).
Los estudios de diagnóstico por imágenes del cuello para la localización de tumores dentro de las glándulas tiroides y paratiroides deben comenzar con ecografía seguida de TC, RM o gammagrafía con radioisótopos según sea necesario. La tomografía computarizada (TC) o la resonancia magnética (RM) son útiles para localizar el feocromocitoma o confirmar la presencia de lesiones bilaterales.
En numerosas ocasiones, el cuadro se identifica durante la evaluación sistemática de miembros de una familia con la enfermedad. También debe sospecharse una NEM 2A en pacientes con feocromocitoma bilateral o con al menos 2 de sus manifestaciones endocrinológicas características. El diagnóstico puede confirmarse con análisis genético. Aunque solo el 25% de los carcinomas medulares de tiroides son casos familiares, deben realizarse pruebas genéticas en todos los pacientes con esta enfermedad. Las mutaciones de RET se encuentran en casi todos los pacientes con cáncer de tiroides medular familiar, pero su incidencia es tan alta como aproximadamente del 7% en casos aparentemente esporádicos (1).
Cribado genético
La evaluación genética de los miembros de familias con NEM 2A es el método de diagnóstico de elección; la disponibilidad de esta técnica determinó que las pruebas bioquímicas para detectar el carcinoma medular de tiroides en forma temprana sean obsoletas. La mutación RET específica también predice características fenotípicas como la agresividad del carcinoma medular de tiroides y la presencia de otras endocrinopatías, por lo que es importante en el manejo clínico; sin embargo, otros factores, como la edad avanzada al inicio y el estadio tumoral más alto en el momento del diagnóstico, pueden ser más predictivos de la agresividad de la enfermedad (2).
El diagnóstico genético preimplantacional y la biopsia de vellosidades coriónicas o amniocentesis prenatal se han utilizado para el diagnóstico prenatal.
En pacientes con miembros de la familia afectados, el cribado anual para hiperparatiroidismo y feocromocitoma debe comenzar en la adolescencia (a los 11 años para las familias con variantes de alto riesgo y a los 16 años para aquellos con variantes de riesgo moderado) y continuar indefinidamente. La detección del hiperparatiroidismo implica la medición de los niveles séricos de calcio. La detección del feocromocitoma incluye preguntas sobre los síntomas, la medición de la frecuencia del pulso y la tensión arterial, y las pruebas de laboratorio.
Referencias del diagnóstico
1. Wells SA Jr, Asa SL, Dralle H, et al: American Thyroid Association Guidelines Task Force on Medullary Thyroid Carcinoma. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid 25(6):567–610, 2015.
2. Voss RK, Feng L, Lee JE, et al: Medullary thyroid carcinoma in MEN2A: ATA moderate- or high-risk RET mutations do not predict disease aggressiveness. J Clin Endocrinol Metab 102(8):2807–2813, 2017.
Tratamiento de la NEM 2A
Extirpación quirúrgica de los tumores identificados
Tiroidectomía profiláctica
En los pacientes con feocromocitoma y carcinoma medular de tiroides o hiperparatiroidismo, primero debe extirparse el feocromocitoma, incluso aunque sea asintomático porque este tumor aumenta en gran medida el riesgo durante otras cirugías (1). Los pacientes sometidos a resección de un feocromocitoma deben recibir un bloqueo alfa adecuado (típicamente con fenoxibenzamina, doxazosina o prazosina) antes de la cirugía. La suprarrenalectomía laparoscópica, que tiene menor morbilidad, es preferible a la laparotomía abierta. Debido a que los feocromocitomas bilaterales son comunes, la cirugía con preservación suprarrenal puede ser apropiada en algunos pacientes (2).
La cirugía para el carcinoma medular de tiroides debe incluir tiroidectomía total y vaciamiento ganglionar del compartimento central, con disección adicional de los ganglios linfáticos, si se indica, según las imágenes preoperatorias. La evaluación posquirúrgica para la enfermedad residual o recurrente debe incluir la medición de la calcitonina sérica y las imágenes con ecografía de cuello y, cuando esté indicado, una tomografía computarizada o una resonancia magnética de cuello y tórax, gammagrafía ósea o tomografía por emisión de positrones (PET).
Una vez que el carcinoma medular de tiroides ha metastatizado, los inhibidores de la tirosina cinasa, incluyendo selpercatinib, cabozantinib y vandetanib recientemente disponibles, pueden alargar la supervivencia libre de progresión. Están en curso ensayos clínicos que evalúan otros inhibidores de la tirosina cinasa para el carcinoma medular de tiroides metastásico (3) La quimioterapia citotóxica y la radioterapia son poco eficaces para prolongar la supervivencia, pero pueden reducir la velocidad de progresión de la enfermedad. Se debe considerar la radioterapia externa adyuvante posoperatoria en pacientes con alto riesgo de recurrencia local y en aquellos con riesgo de obstrucción de la vía aérea. Algunos estudios han demostrado prolongar la supervivencia con inmunoterapia (p. ej., vacunas derivadas de tumores, células tumorales transfectadas) y radioinmunoterapia (p. ej., anticuerpos monoclonales acoplados a radioisótopos).
Cuando la evaluación genética identifica a un niño con la mutación en RET, se recomienda la tiroidectomía profiláctica. Dependiendo de la mutación particular, puede estar indicada la tiroidectomía profiláctica desde los primeros meses de vida. El cáncer medular de tiroides puede curarse o prevenirse mediante una tiroidectomía temprana. Existe una correlación genotipo-fenotipo para los pacientes con mutaciones RET que puede proporcionar información sobre la edad de inicio y el curso clínico del cáncer medular de tiroides, lo que es determinante para definir el momento de la cirugía en un niño afectado.
La angustia psicológica parece ser común y crónica en pacientes con NEM 2. Los factores contribuyentes incluyen poca cantidad de información sobre la enfermedad, tener hijos con la mutación, número de cirugías y presencia de comorbilidades; se recomienda una evaluación psicológica para identificar y tratar a las personas afectadas (4).
Referencias del tratamiento
1. Wells SA Jr: Advances in the management of MEN2: From improved surgical and medical treatment to novel kinase inhibitors. Endocr Relat Cancer 25:T1–T13, 2018.
2. Castinetti F, Qi XP, Walz AL, et al: Outcomes of adrenal-sparing surgery or total adrenalectomy in phaeochromocytoma associated with multiple endocrine neoplasia type 2: An international retrospective population-based study. Lancet Oncol 15(6):648–655, 2014.
3. Okafor C, Hogan J, Raygada M, et al: Update on Targeted Therapy in Medullary Thyroid Cancer. Front Endocrinol (Lausanne). 12:708949, 2021. doi:10.3389/fendo.2021.708949
4. Rodrigues KC, Toledo RA, Coutinho FL, et al: Assessment of depression, anxiety, quality of life, and coping in long-standing multiple endocrine neoplasia type 2 patients. Thyroid 27(5):693–706, 2017.
Conceptos clave
La mayoría de los pacientes con neoplasia endocrina múltiple tipo 2A presentan un carcinoma medular de la glándula tiroides, que por lo general se inicia en la infancia.
Otras manifestaciones son las del exceso de hormonas, especialmente la hipertensión secundaria a feocromocitoma y la hipercalcemia generada por hiperparatiroidismo.
Los pacientes deben someterse a pruebas genéticas para las mutaciones del protooncogén RET y evaluación clínica para otros tumores del síndrome.
Los tumores se extirpan cuando sea posible, empezando por los feocromocitomas.
Se recomienda la tiroidectomía profiláctica; el momento de la cirugía puede estar influido por la mutación específica.
Más información
El siguiente recurso en inglés puede ser útil. Tenga en cuenta que el MANUAL no es responsable por el contenido de este recurso.
Saravana-Bawan B, Pasternak JD. Multiple endocrine neoplasia 2: an overview. Ther Adv Chronic Dis 2022;13:20406223221079246. Publicado el 25 de febrero de 2022. doi:10.1177/20406223221079246









