



Pharmacokinetics refers to the processes of drug absorption, distribution, metabolism, and elimination. There are important age-related variations in pharmacokinetics.
Absorption
Absorption from the gastrointestinal tract is affected by:
Gastric acid secretion
Bile salt formation
Gastric emptying time
Intestinal motility
Bowel length and effective absorptive surface
Intestinal microbial flora
Illness
All the above factors vary with age (1).
Reduced gastric acid secretion increases the bioavailability of acid-labile drugs (eg, penicillin) and decreases bioavailability of weakly acidic drugs (eg, phenobarbital) (2). Reduced bile salt formation decreases the bioavailability of lipophilic drugs (eg, diazepam).
Reduced gastric emptying and intestinal motility increase the time taken to reach therapeutic concentrations when enteral drugs are given to infants < 3 months old. Drug-metabolizing enzymes present in the intestines of young infants are another cause of reduced drug absorption. Infants with congenital atretic bowel or surgically removed bowel or who have jejunal feeding tubes may have specific absorptive defects depending on the length of bowel lost or bypassed and the location of the lost segment. How the type of food consumed may alter gastric emptying should also be considered (eg, solid versus liquid).
Intestinal flora may aid in metabolism of some medications such as digoxin. Changes in the type or amount of intestinal flora may affect absorption and efficacy (2).
Injected medications are often erratically absorbed because of:
Variability in their chemical characteristics
Differences in absorption by site of injection (intramuscular or subcutaneous)
Variability in muscle mass among children
Illness (eg, compromised circulatory status)
Variability in depth of injection (too deep or too shallow)
Skeletal muscle blood flow and muscular contractions
Intramuscular injections are generally avoided in children because of pain and the possibility of tissue damage, but, when needed, water-soluble drugs are best because they do not form precipitates at the injection site.
Transdermal absorption may be enhanced in neonates and young infants because the stratum corneum is thin and because the ratio of surface area to weight is much greater than for older children and adults. Skin disruptions (eg, abrasions, eczema, burns) increase absorption in children of any age. Caution should be used for topically applied drugs such as glucocorticoids, as there can be increased systemic exposure and absorption, particularly in neonates.
Transrectal drug therapy is generally appropriate only for emergencies when an IV route is not available (eg, use of rectal diazepam for status epilepticus). The site of placement of the drug within the rectal cavity may influence absorption because of the difference in venous drainage systems. Young infants may also expel solid drugs before significant absorption has occurred.
Absorption of inhaled drugs from the lungs (eg, beta-agonists for asthma, pulmonary surfactant for respiratory distress syndrome) may be influenced more by the reliability of the delivery device and the technique used by the patient or caregiver than by physiologic parameters.
Absorption references
1. van den Anker J, Reed MD, Allegaert K, Kearns GL: Developmental changes in pharmacokinetics and pharmacodynamics. J Clin Pharmacol 58 (supplement 10):S10–S25, 2018. doi: 10.1002/jcph.1284
2. Kearns GL, Abdel-Rahman SM, Alander SW, Blowey DL, Leeder JS, Kauffman RE: Developmental pharmacology--drug disposition, action, and therapy in infants and children. N Engl J Med 349(12):1157–1167, 2003. doi:10.1056/NEJMra035092
Distribution
The volume of distribution of drugs changes in children as they age. These age-related changes are due to changes in body composition (especially the extracellular space and total body water content), plasma protein binding, and inherent drug characteristics (eg, water or lipid solubility, ionization).
Higher doses (per kg of body weight) of water-soluble drugs are required in younger children because a higher percentage of their body weight is water (see figure ). Conversely, lower doses are required to avoid toxicity as children grow older because of the decline in water as a percentage of body weight. Additionally, children with obesity have been shown to have significantly higher percentages of total body water, body volume, lean body mass, and fat mass compared to children without obesity (1).
Changes in Body Composition With Growth and Aging
Adapted from Puig M: Body composition and growth. In Nutrition in Pediatrics, ed. 2, edited by WA Walker and JB Watkins. Hamilton, Ontario, BC Decker, 1996. |

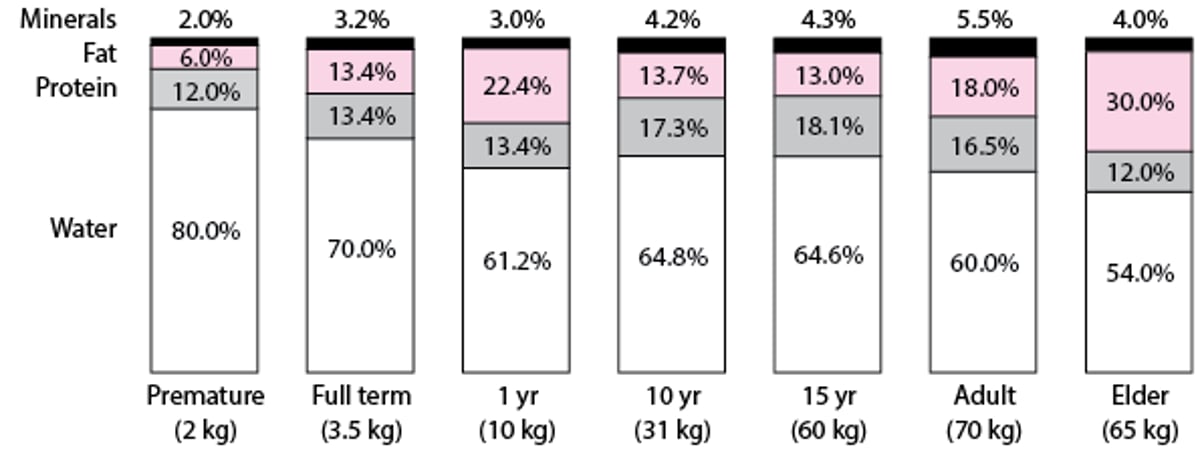
Many drugs bind to proteins, primarily albumin, alpha-1 acid glycoprotein, and lipoproteins; protein-binding limits the distribution of free drug throughout the body. Albumin and total protein concentrations are lower in neonates but approach adult levels by 10 to 12 months. Decreased protein-binding in neonates is also due to qualitative differences in binding proteins (ie, the presence of fetal albumin) and to competitive binding by molecules such as bilirubin and free fatty acids, which circulate in higher concentrations in neonates and infants. The net result may be increased free drug concentrations, greater drug availability at receptor sites, and both increased pharmacologic effects and higher frequency of adverse effects at lower drug concentrations.
Distribution reference
1. Vaughns JD, Conklin LS, Long Y, et al: Obesity and pediatric drug development. J Clin Pharmacol 58(5):650–661, 2018. doi: 10.1002/jcph.1054
Metabolism and elimination
Drug metabolism and elimination vary with age and depend on the substrate or drug, but most drugs (notably phenytoin, barbiturates, analgesics, and cardiac glycosides) have plasma half-lives 2 to 3 times longer in neonates than in adults.
The cytochrome P-450 (CYP450) enzyme system in the small bowel and liver is the most important known system for drug metabolism. CYP450 enzymes inactivate drugs via:
Oxidation, reduction, and hydrolysis (phase I metabolism)
Hydroxylation and conjugation (phase II metabolism)
Phase I metabolism activity is reduced in neonates, increases progressively during the first 6 months of life, exceeds adult rates by the first few years for some drugs, slows during adolescence, and usually attains adult rates by late puberty. However, adult rates of metabolism may be achieved for some drugs (eg, barbiturates, phenytoin) 2 to 4 weeks postnatally. CYP450 activity can also be induced (reducing drug concentrations and effect) or inhibited (augmenting concentrations and effect) by coadministered drugs. Drug interactions between coadministered drugs may lead to drug toxicity when CYP450 activity is inhibited or to an inadequate drug level when CYP450 activity is induced. Diet may also affect development of CYP450 activity in children, as evidenced by differences in CYP1A2 and CYP3A4 activity in infants who are fed formula, as compared to infants who are breastfed (1). The kidneys, lungs, and skin also play a role in the metabolism of some drugs, as do intestinal drug-metabolizing enzymes in neonates.
Phase II metabolism varies considerably by substrate. Maturation of enzymes responsible for bilirubin and acetaminophen conjugation is delayed; enzymes responsible for morphine conjugation are fully mature even in preterm infants.
Drug metabolites are eliminated primarily through bile or urine (ie, via the kidneys). Renal elimination depends on:
Plasma protein binding
Renal blood flow
Glomerular filtration rate
Tubular secretion
All of these factors are altered in the first 2 years of life. Renal plasma flow is low at birth (12 mL/minute) and reaches adult levels of 140 mL/minute by age 1 year. Similarly, glomerular filtration rate is 2 to 4 mL/minute at birth (rates are lower in preterm neonates), increases to 8 to 20 mL/minute by 2 to 3 days, and reaches adult levels of 120 mL/minute by 8 to 12 months (2). Tubular secretion also matures during the first few years of life, and the ability of the renal tubules to secrete electrolytes and drugs improves as the child grows, reaching adult levels by the first year of life.
Metabolism and elimination references
1. Blake JB, Abdel-Rahman SM, Pearce RE, et al: Effect of diet on the development of drug metabolism by cytochrome P-450 enzymes in healthy infants. Pediatr Res 60(6):717–723, 2006. doi: 10.1203/01.pdr.0000245909.74166.00
2. Kearns GL, Abdel-Rahman SM, Alander SW, Blowey DL, Leeder JS, Kauffman RE: Developmental pharmacology--drug disposition, action, and therapy in infants and children. N Engl J Med 349(12):1157–1167, 2003. doi:10.1056/NEJMra035092
Drug dosing
Because of the above factors, drug dosing in children < 12 years old is frequently a function of age, body weight, or both. This approach is practical but not ideal. Even within a population of similar age and weight, drug requirements may differ because of maturational differences in absorption, distribution, metabolism, and elimination. Thus, when practical, dose adjustments should be based on plasma drug concentration (noting that plasma drug concentration may not reflect the drug concentration in the target organ). Unfortunately, these adjustments are not feasible for most drugs. However, in the United States, because of the Best Pharmaceuticals for Children Act of 2001 and the Pediatric Research Equity Act of 2003 (both made permanent in 2012 [1]), more complete pediatric dosing, pharmacokinetic, and safety information is available for over 900 drugs for use in children (2).
Physiologically based pharmacokinetic modeling is a mathematical technique that uses known principles of biochemistry and physiology to predict how a drug will be absorbed, distributed, metabolized, and excreted. Results of this modeling can help support decisions on whether, when, and how to conduct a clinical trial and can help improve the safety and efficiency of pediatric clinical trials.
Drug dosing references
1. Bourgeois FT, Kesselheim AS: Promoting pediatric drug research and labeling—Outcomes of legislation. N Engl J Med 381(9):875–881, 2019. doi: 10.1056/NEJMhle1901265
2.U.S. Food and Drug Administration (FDA): Best Pharmaceuticals for Children Act and Pediatric Research Equity Act status report (2020). Accessed June 4, 2025
Drug Information for the Topic





