

Fabry disease is a sphingolipidosis, an inherited disorder of metabolism, caused by deficiency of alpha-galactosidase A, which causes angiokeratomas, acroparesthesias, corneal opacities, recurrent febrile episodes, and renal or heart failure. Diagnosis is clinical and by DNA analysis and/or enzyme analysis of amniocytes or chorionic villi or, postnatally, of serum or white blood cells. Treatment is enzyme replacement with recombinant alpha-galactosidase A or migalastat.A or migalastat.
For more information, see table .
See also Approach to the Patient With a Suspected Inherited Disorder of Metabolism.
Fabry disease is an X-linked deficiency of the lysosomal enzyme alpha-galactosidase A, which is needed for normal trihexosylceramide catabolism. Glycolipid (globotriaosylceramide) accumulates in many tissues (eg, vascular endothelium, lymph vessels, heart, kidney).
Diagnosis of Fabry Disease
Clinical evaluation
Enzyme analysis
Sometimes DNA analysis
Diagnosis in males is by enzyme analysis and is clinical based on appearance of typical skin lesions (angiokeratomas) over the lower trunk and by characteristic features of peripheral neuropathy (causing recurrent burning pain in the extremities), corneal opacities, and recurrent febrile episodes. Death results from renal failure or cardiac or cerebral complications of hypertension or other vascular disease.
Heterozygous females are usually asymptomatic but may have an attenuated form of disease often characterized by corneal opacities.

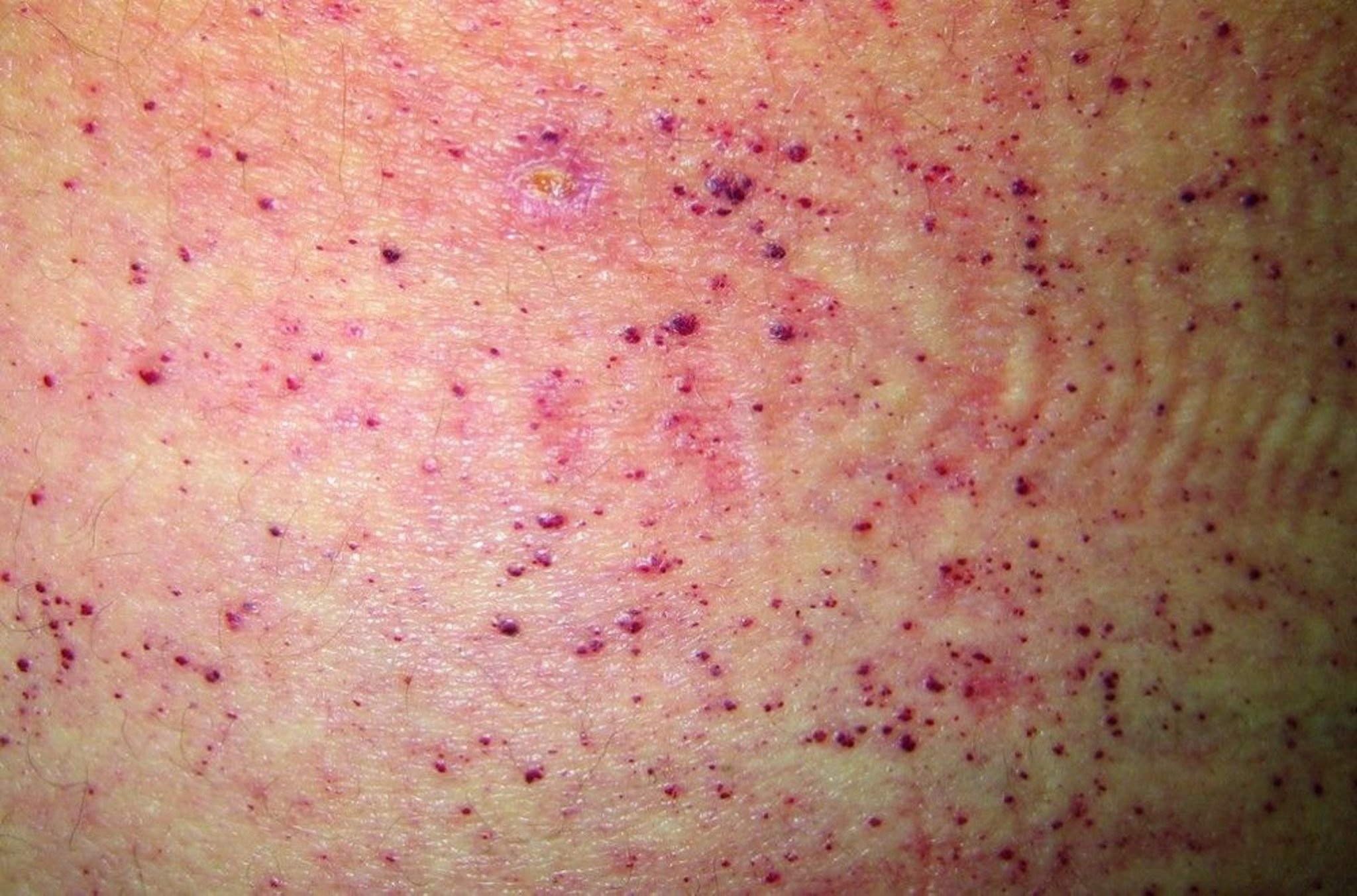
© Springer Science+Business Media
Diagnosis of Fabry disease is by DNA analysis (especially in females) and/or assay of galactosidase activity—prenatally in amniocytes or chorionic villi and postnatally in serum or white blood cells. (See also testing for suspected inherited disorders of metabolism.)
Treatment of Fabry Disease
Enzyme replacement
Sometimes migalastat
Treatment is enzyme replacement with recombinant alpha-galactosidase A (agalsidase alpha or agalsidase beta) combined with supportive measures for fever and pain (Treatment is enzyme replacement with recombinant alpha-galactosidase A (agalsidase alpha or agalsidase beta) combined with supportive measures for fever and pain (1).
Migalastat, an oral pharmacologic chaperone that binds to and stabilizes specific mutant forms of Migalastat, an oral pharmacologic chaperone that binds to and stabilizes specific mutant forms ofalpha-galactosidase, is used as an alternative in some patients.
Kidney transplantation is effective for treating renal failure.
Treatment reference
1. Germain DP, Altarescu G, Barriales-Villa R, et al. An expert consensus on practical clinical recommendations and guidance for patients with classic Fabry disease. Mol Genet Metab. 2022;137(1-2):49-61. doi:10.1016/j.ymgme.2022.07.010
More Information
The following English-language resource may be useful. Please note that THE MANUAL is not responsible for the content of this resource.
Online Mendelian Inheritance in Man (OMIM) database: Complete gene, molecular, and chromosomal location information





