

Vasculitis is characterized by inflammation of blood vessels, which can lead to vessel wall destruction and downstream tissue ischemia and necrosis. Vasculitis may be associated with organ inflammation, but inflammation may not be from vasculitis. Vasculitis can affect any blood vessel—arteries, arterioles, veins, venules, or capillaries. Clinical manifestations of specific vasculitic disorders are diverse and depend on the size and location of the involved vessels, the extent of the organ involvement, and the degree and pattern of extravascular inflammation.
Etiology of Vasculitis
Vasculitis may be
Primary
Secondary
Primary vasculitis has no known cause.
Secondary vasculitis may be triggered by an infection, a medication, or a toxin, or may occur as part of another inflammatory disorder (eg, systemic lupus erythematosus, relapsing polychondritis) or cancer.
Pathophysiology of Vasculitis
Histologic description of an affected vessel should include the following:
A description of vessel wall damage (eg, type and location of inflammatory infiltrate, extent and type of damage, presence or absence of fibrinoid necrosis)
A description of healing responses (eg, intimal thickening, fibrosis)
Certain features (eg, predominant inflammatory cell type, location of inflammation) suggest particular vasculitic processes and may aid in the diagnosis. For example, in many acute lesions, the predominant inflammatory cells are polymorphonuclear leukocytes; in chronic lesions, lymphocytes may predominate.
Inflammation may be segmental or involve the entire vessel. At sites of inflammation, varying degrees of cellular inflammation and necrosis or scarring occur in one or more layers of the vessel wall. Inflammation in the media of a muscular artery tends to destroy the internal elastic lamina. Some forms of vasculitis are characterized by giant cells in the vessel wall. In some vasculitic disorders, such as granulomatosis with polyangiitis or Kawasaki disease, the vessel inflammation (true vasculitis) is only part of the pathophysiology and there is predominant parenchymal inflammation in a characteristic pattern that involves specific organs.
Leukocytoclastic vasculitis is a histopathologic term used to describe findings in cutaneous small-vessel vasculitis. It refers to breakdown of inflammatory cells that leaves small nuclear fragments (nuclear debris) in and around the vessels. Inflammation is transmural and nongranulomatous. Polymorphonuclear leukocytes predominate early; later, lymphocytes predominate. Resolution of the inflammation tends to result in fibrosis and intimal thickening. Intimal thickening or secondary clot formation can narrow the vessel lumen, decrease blood flow, and cause tissue ischemia or necrosis.
Histologic Clues to Diagnosis of Vasculitic Disorders
Findings | Possible Diagnoses |
|---|---|
Predominantly nonnecrotizing granulomatous inflammatory infiltrate with lymphocytes, macrophages, and multinucleated giant cells | Primary angiitis of the central nervous system (certain types) |
Fibrinoid vascular necrosis of the vessel wall with a mixed infiltrate consisting of various combinations of leukocytes and lymphocytes | Eosinophilic granulomatosis with polyangiitis (EGPA) Granulomatosis with polyangiitis (GPA) Immune complex–associated vasculitis |
IgA deposits* | Immunoglobulin A–associated vasculitis (formerly Henoch-Schönlein purpura) |
Scant or complete absence of immunoglobulins and complement deposition in the vessel walls*† | EGPA GPA MPA |
* These observations are noted using immunofluorescence staining. | |
† Disorders thus characterized are called pauci-immune vasculitic disorders. | |
Classification of Vasculitis
Vasculitic disorders can be classified according to the size of the predominant vessel affected. However, there is often substantial overlap. Nomenclature has been developed to help classify the various disorders by predominant vessel involvement (ie, large-vessel, medium-vessel, and small-vessel) and disease category when relevant (1, 2). Single-organ vasculitides refers to a vasculitis that is limited to a single organ (eg, primary central nervous system vasculitis).
Classification of Vasculitic Disorders
Category | Disorders |
|---|---|
Large-vessel vasculitis | |
Medium-vessel vasculitis | |
Small-vessel vasculitis | Antineutrophil cytoplasmic antibody (ANCA)–associated vasculitis (AAV)
Immune complex small vessel vasculitis
|
Variable-vessel vasculitis | Cogan syndrome |
Single-organ vasculitis | Cutaneous small vessel vasculitis Primary central nervous system vasculitis Isolated aortitis |
Data from Jennette JC, Falk RJ, Bacon PA, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum 2013;65(1):1-11. doi:10.1002/art.37715 | |
Classification references
1. Jennette JC, Falk RJ, Bacon PA, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum 2013;65(1):1-11. doi:10.1002/art.37715
2. Sunderkötter CH, Zelger B, Chen KR, et al. Nomenclature of Cutaneous Vasculitis: Dermatologic Addendum to the 2012 Revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheumatol 2018;70(2):171-184. doi:10.1002/art.40375
Symptoms and Signs of Vasculitis
Size of the affected vessels helps determine clinical presentation.
Regardless of the size of the vessels involved, patients can present with symptoms and signs of systemic inflammation (eg, fever, night sweats, fatigue, anorexia, weight loss, arthralgias, arthritis). Some manifestations are life-threatening or organ-threatening and require immediate treatment:
Small- and medium-sized vasculitides often manifest with skin lesions such as palpable purpura, urticaria, ulcers, livedo reticularis, and nodules.

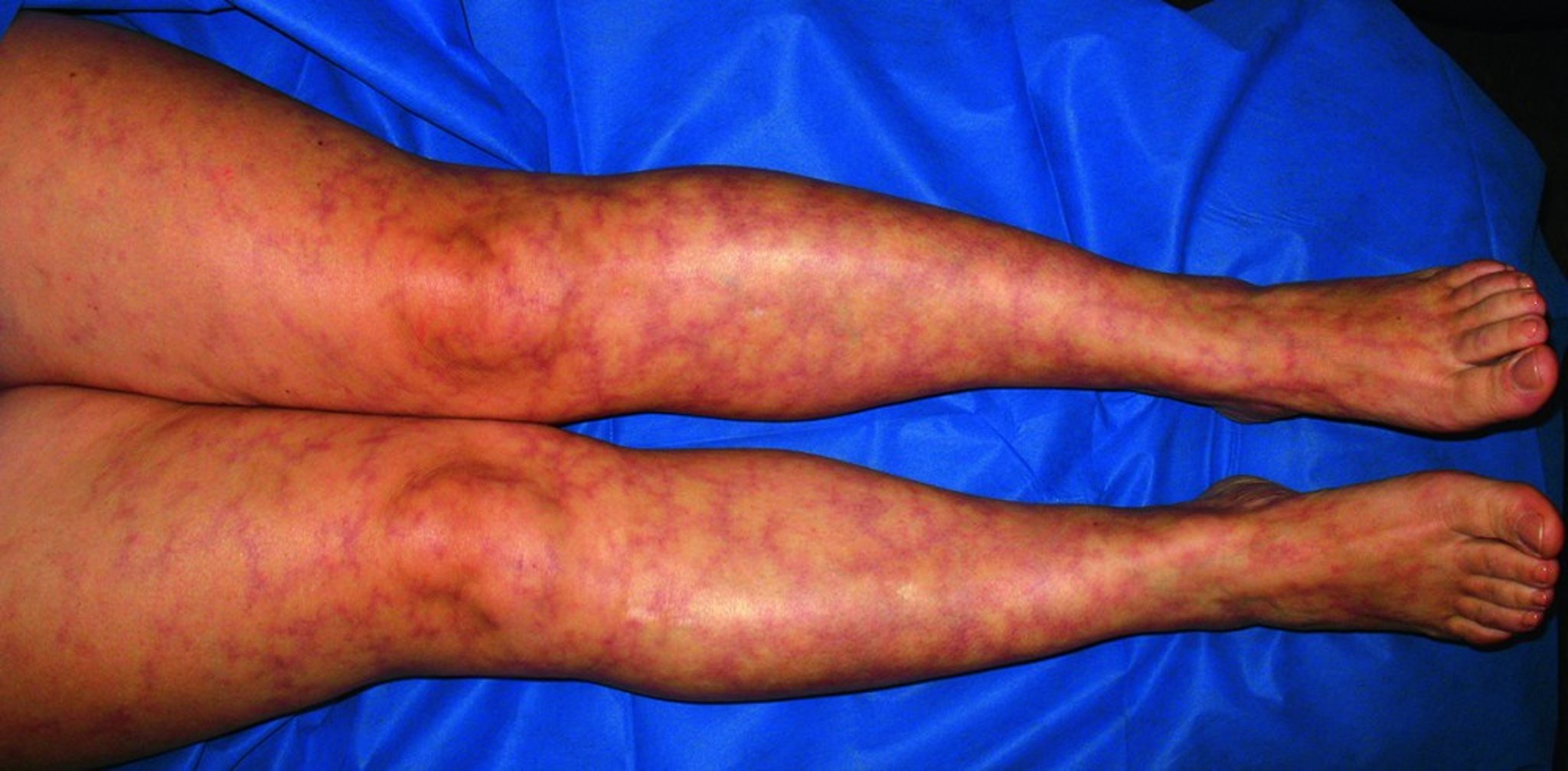
This photo shows the lacy pattern of erythema typical of livedo reticularis.
© Springer Science+Business Media

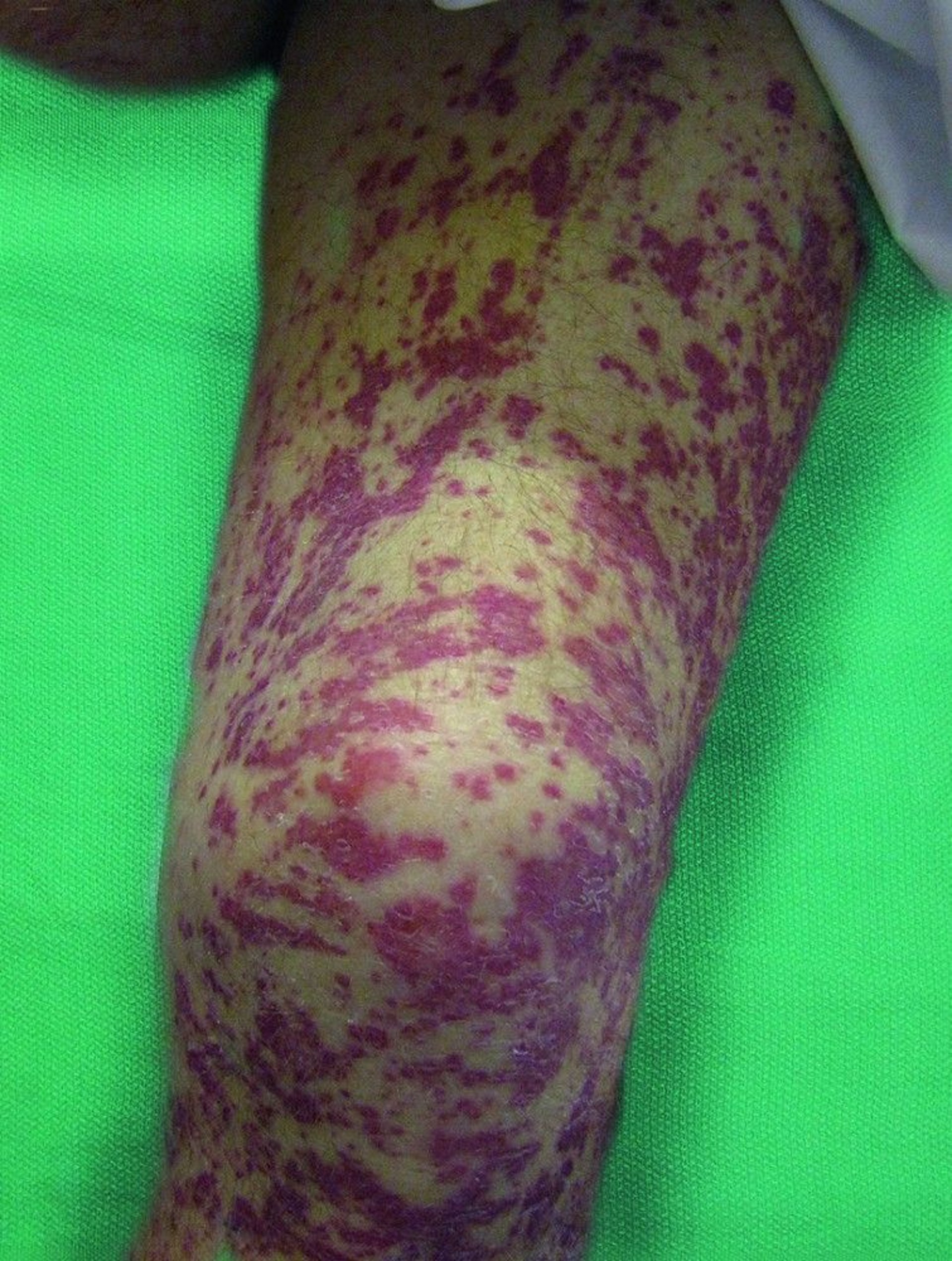
This image shows multiple ecchymoses and purpura on the legs.
© Springer Science+Business Media
Diagnosis of Vasculitis
History and physical examination
Basic laboratory tests to detect inflammation and organ dysfunction (eg, complete blood count [CBC], erythrocyte sedimentation rate [ESR] or C-reactive protein, serum albumin and total protein, aspartate aminotransferase [AST] and alanine aminotransferase [ALT], blood urea nitrogen [BUN] and creatinine, urinalysis) and to stage disease process
Laboratory tests to help determine the type of vasculitis (eg, antineutrophil cytoplasmic antibodies [ANCA]) if suggested by clinical assessment
Laboratory and imaging studies that may help determine the cause of vasculitis (eg, cryoglobulins, hepatitis B surface antigen test, hepatitis B core and hepatitis B surface antibody tests, hepatitis C virus antibody test, blood cultures) and extent of organ involvement
Biopsy
Systemic vasculitis is suspected in patients with the following:
Symptoms or signs suggestive of vasculitis (eg, temporal headache and jaw claudication suggesting giant cell arteritis)
Manifestations of ischemia (eg, ischemic stroke, limb claudication, mesenteric ischemia) out of proportion to a patient's risk factors for atherosclerosis
Unexplained combinations of symptoms in more than one organ system that are compatible with vasculitis (eg, hypertension, myalgias, hemoptysis), particularly when symptoms of a systemic illness are present
Primary vasculitic disorders are diagnosed based on the presence of characteristic symptoms, physical findings, compatible laboratory test results, and exclusion of other causes (ie, secondary vasculitis). Histologic examination is done whenever possible and may support the diagnosis of a particular vasculitic disorder (see table ). Clinical findings determine the differential diagnosis and thus direct laboratory testing.
Most routine laboratory tests yield results that are nonspecific and must be interpreted in the context of the entire clinical presentation. However, results can often help support the diagnosis, determine the location and degree of organ involvement, or suggest alternative diagnoses. Tests usually include CBC, ESR and/or C-reactive protein, serum albumin and total protein, AST, and ALT. Often, patients present with elevated ESR or C-reactive protein, anemia due to chronic inflammation, elevated platelets, and low serum albumin. Freshly voided urine must be tested for red blood cells, red blood cell casts, and protein to identify renal involvement. Serum creatinine levels should be checked and monitored. Leukopenia and thrombocytopenia are not typical of primary vasculitis and suggest an alternate diagnosis.
Detection of antineutrophil cytoplasmic antibodies (ANCA) may support the diagnosis of granulomatosis with polyangiitis (GPA), eosinophilic granulomatosis with polyangiitis (EGPA), or microscopic polyangiitis (collectively called ANCA-associated vasculitides). Standardized tests for ANCA include immunofluorescence staining and enzyme-linked immunosorbent assay (ELISA). Immunofluorescence staining of ethanol-fixed neutrophils can detect the cytoplasmic pattern of c-ANCA or the perinuclear pattern of p-ANCA. Then ELISA is used to check for antibodies specific for the major autoantigens: proteinase-3 (PR3), which produces the c-ANCA staining pattern, or myeloperoxidase (MPO), which produces the p-ANCA staining pattern seen on ethanol-fixed neutrophils. Because ANCA-associated vasculitides are rare, and the ANCA test is not completely specific, ANCA testing should be done only when the pretest probability for ANCA-associated vasculitis is moderately high. A positive ANCA test can occur in infections that can cause a secondary vasculitis, including endocarditis.
Other useful laboratory tests include hepatitis B and hepatitis C serologic testing, serum and urine protein electrophoresis, antinuclear antibody and anti-extractable nuclear antigens panel, testing for the presence of cryoglobulins, and complement levels. Complement levels may be low in viral vasculitis, cryoglobulinemic vasculitis, lymphoproliferative disorders, or vasculitis secondary to other autoimmune diseases.
Further testing is determined by clinical findings. If indicated based on clinical findings, a chest radiograph should be done to check for infiltrates, but high-resolution noncontrast CT of the chest may be needed to check for subtle findings, such as small nodules or cavities. Bilateral diffuse infiltrates suggest possible alveolar hemorrhage, which requires immediate diagnosis and treatment. Other imaging tests may be required. For example, magnetic resonance angiography of large blood vessels and the aorta is useful for diagnosis and monitoring when such vessels appear affected. If symptoms and examination suggest a neuropathy, electromyography may be helpful.
Because vasculitic disorders are rare and treatment may have severe adverse effects, tissue biopsy is done to confirm the diagnosis whenever possible. Clinical findings suggest the best site for biopsy. Biopsy results are most likely to be positive if taken from affected lung, skin, and kidney tissue. Biopsies of organs without clinical manifestations or laboratory suggestion of involvement have a low likelihood of providing positive results.
Treatment of Vasculitis
Induction of remission with corticosteroids, often with another immunosuppressant in most forms of vasculitis (eg, rituximab for ANCA-associated vasculitis)Induction of remission with corticosteroids, often with another immunosuppressant in most forms of vasculitis (eg, rituximab for ANCA-associated vasculitis)
Maintenance of remission usually with tapering of corticosteroids to reduce toxicity plus another immunosuppressant (eg, rituximab for ANCA-associated vasculitis)Maintenance of remission usually with tapering of corticosteroids to reduce toxicity plus another immunosuppressant (eg, rituximab for ANCA-associated vasculitis)
Treatment of vasculitis depends on the etiology, the type of vasculitis, and the extent and severity of disease. For secondary vasculitic disorders, treating the underlying disease (eg. for cryoglobulinemic vasculitis associated with hepatitis C) or removing the cause (eg, infection, medication, cancer) usually helps.
For primary vasculitic disorders, treatment aims to induce and maintain remission. The duration of remission is hard to predict and may depend on the type of vasculitis. For many patients, maintaining remission requires continuation of immunosuppressive therapy with or without a low dose of corticosteroids. During this period, the goal is to eliminate corticosteroids or reduce their dose and use alternative, less toxic immunosuppressants as long as needed.
All patients treated with immunosuppressants should be monitored for opportunistic and other infections. Testing for tuberculosis, hepatitis B, and hepatitis C, which can become reactivated or exacerbated by some immunosuppressive therapies, should be done. Prophylaxis against Pneumocystis jirovecii should be considered for patients receiving potent or prolonged immunosuppressive therapy.
Induction of remission
In most forms of vasculitis, initial treatment begins with corticosteroids, and an additional immunosuppressant. Treatment approach depends on disease severity. In general:
Severe, rapidly progressive and life- or organ-threatening vasculitis (eg, causing alveolar hemorrhage, rapidly progressive glomerulonephritis, or mesenteric ischemia) is a medical emergency requiring hospital admission and immediate treatment. High-dose corticosteroids are typically prescribed. Specific doses must be individualized. For example, for severe ANCA-associated vasculitis, methylprednisolone 1 g IV once a day for 3 days ("pulse dose") may be used, followed by 1 mg/kg of prednisone (or the equivalent dose of methylprednisolone) orally once a day. The dose is then tapered slowly, as tolerated, until the medication is stopped. Changes in tapering schedule may be necessary if the patient fails to improve or relapses. Rituximab (or cyclophosphamide) is often initiated with corticosteroids for induction of remission and allow for tapering of the corticosteroid dose.Severe, rapidly progressive and life- or organ-threatening vasculitis (eg, causing alveolar hemorrhage, rapidly progressive glomerulonephritis, or mesenteric ischemia) is a medical emergency requiring hospital admission and immediate treatment. High-dose corticosteroids are typically prescribed. Specific doses must be individualized. For example, for severe ANCA-associated vasculitis, methylprednisolone 1 g IV once a day for 3 days ("pulse dose") may be used, followed by 1 mg/kg of prednisone (or the equivalent dose of methylprednisolone) orally once a day. The dose is then tapered slowly, as tolerated, until the medication is stopped. Changes in tapering schedule may be necessary if the patient fails to improve or relapses. Rituximab (or cyclophosphamide) is often initiated with corticosteroids for induction of remission and allow for tapering of the corticosteroid dose.
Less severe presentations of vasculitis may be managed with lower doses of corticosteroids, along with less potent immunosuppressants. For example, for patients with ANCA-associated vasculitis that is not organ-threatening, weekly oral methotrexate may be considered in addition to corticosteroid.Less severe presentations of vasculitis may be managed with lower doses of corticosteroids, along with less potent immunosuppressants. For example, for patients with ANCA-associated vasculitis that is not organ-threatening, weekly oral methotrexate may be considered in addition to corticosteroid.
Remission maintenance
Corticosteroids are tapered off or to the lowest dose that can maintain remission. For several forms of vasculitis, a less toxic immunosuppressant is prescribed to maintain remission as the corticosteroids are decreased and ideally eliminated. The duration of maintenance therapy varies, from 1 year to several years, depending on the patient, the specific diagnosis, and the propensity for relapse. Patients with frequent relapses may need to take immunosuppressants indefinitely.
Long-term use of corticosteroids can have significant adverse effects. Patients who are taking ≥ 7.5 mg of prednisone daily or equivalent doses of other corticosteroids should be given calcium and vitamin D supplements and bisphosphonates to help Long-term use of corticosteroids can have significant adverse effects. Patients who are taking ≥ 7.5 mg of prednisone daily or equivalent doses of other corticosteroids should be given calcium and vitamin D supplements and bisphosphonates to helpprevent or minimize osteoporosis; bone density monitoring should be considered. Additional corticosteroids may be needed in patients who are seriously ill or in those undergoing surgery who may have a suppressed hypothalamic-pituitary-adrenal axis, depending on the dose and duration of corticosteroid therapy and the duration and intensity of stress.
Key Points
Vasculitis can be a primary disorder or secondary to other causes.
Vasculitis tends to affect small-, medium-, or large-sized vessels, each with certain patterns of organ involvement.
Clinical manifestations can be systemic and/or organ-specific, depending on the vessels that are affected.
Perform blood tests, imaging studies, and tissue biopsy as indicated to determine the cause of vasculitis (including disorders such as infections and cancer), extent of organ involvement, and severity of disease.
Treat with corticosteroids and other immunosuppressants.
Address increased risks of infection and osteoporosis caused by vasculitis treatment with monitoring and/or prophylactic treatments.
Drug Information for the Topic





