




先天性心疾患は,最も頻度の高い先天奇形であり,出生児の1%近くに発生する(1)。先天異常のうち,先天性心疾患は乳児期死亡の主要な原因である。
乳児期に診断される最も頻度の高い先天性心疾患は,筋性部および膜性部心室中隔欠損症であり,それに二次孔型心房中隔欠損症が続き,これらを合わせた有病率は出生10,000人当たり48.4例である。最も頻度の高いチアノーゼ性先天性心疾患はファロー四徴症であり,その有病率は大血管転位の2倍である(出生10,000人当たり4.7例 vs. 2.3例)。全体として見ると,大動脈二尖弁が最も頻度の高い先天異常であり,その有病率は0.5~2.0%にも上ると報告されている(2, 3)。
総論の参考文献
1.Reller MD, Strickland MJ, Riehle-Colarusso T, et al: Prevalence of congenital heart defects in metropolitan Atlanta, 1998–2005.J Pediatr 153(6):807–813, 2008.
2.Freeze SL, Landis BJ, Ware SM, Helm BM: Bicuspid aortic valve: a review with recommendations for genetic counseling.J Genet Couns 25(6):1171–1178, 2016.
3.van der Linde D, Konings EEM, Slager MA, et al: Birth prevalence of congenital heart disease worldwide: a systematic review and meta-analysis. J Am Coll Cardiol 58(21):2241–2247, 2011. doi: 10.1016/j.jacc.2011.08.025
病因
先天性心疾患の発生には環境因子と遺伝因子が関与する。
一般的な環境因子としては,母体疾病(例,糖尿病,風疹,全身性エリテマトーデス)や母親による催奇形性物質(例,リチウム,イソトレチノイン,抗てんかん薬)の摂取などがある。母親の年齢は特定の遺伝学的異常(特にダウン症候群)の危険因子として知られており,ダウン症候群には心疾患が含まれることがある。母親の年齢が先天性心疾患の独立した危険因子であるかどうかは明らかでない。父親の年齢も危険因子となりうる(1)。
21トリソミー(ダウン症候群),18トリソミー,13トリソミー,Xモノソミー(ターナー症候群)などの特定の数的染色体異常(異数性)に,先天性心疾患との強い関連が認められる。しかしながら,それらの異常が認められる先天性心疾患の症例は全体の5~6%にすぎない。
その他の症例の多くには,染色体の微細欠失(微小欠失),染色体の微細重複,または単一遺伝子の変異が関与している。しばしば,これらの変異によって,心臓だけでなく複数の臓器が侵される先天性症候群が発生する。その例としては,ディジョージ症候群(22q11.2の微小欠失)やウィリアムズ(またはWilliams-Beuren)症候群(7p11.23の顕微鏡的欠失)などがある。先天性心疾患を合併する症候群を引き起こす単一遺伝子異常としては,フィブリリン-1遺伝子(マルファン症候群)やTXB5(Holt-Oram症候群)などがあり,PTPN11(ヌーナン症候群)変異もその可能性がある。単一遺伝子異常が孤発性の(すなわち症候群を構成しない)先天性心疾患を引き起こすこともある。
先天性心疾患患者の約72%では,同定可能な遺伝学的病因が認められない(2, 3)。
家系内での先天性心疾患の再発生リスクは,その原因に依存する。そのリスクは,de novo変異では無視できるほど小さく,症候群を構成しない多因子性の先天性心疾患では2~5%であり,常染色体優性変異が原因の場合は50%である。家系内有病率が9%と報告されている(4)ことを考慮すると,個々の患者で大動脈二尖弁を同定することは,家族のスクリーニングという観点で有益である。現在では,以前と比べて多くの先天性心疾患患者が成人まで生存し,一部は家族をもつようになってきていることから,遺伝因子を同定することが重要となっている。
病因論に関する参考文献
1.Materna-Kiryluk A, Wiśniewska K, Badura-Stronka M, et al: Parental age as a risk factor for isolated congenital malformations in a Polish population.Paediatr Perinat Epidemiol 23(1):29-40, 2009.doi: 10.1111/j.1365-3016.2008.00979.x
2.Russell MW, Chung WK, Kaltman JR, Miller TA: Advances in the understanding of the genetic determinants of congenital heart disease and their impact on clinical outcomes.J Am Heart Assoc 7(6):e006906, 2018.doi:10.1161/JAHA.117.006906
3.van der Linde D, Konings EEM, Slager MA, et al: Birth prevalence of congenital heart disease worldwide: a systematic review and meta-analysis. J Am Coll Cardiol 58(21):2241–2247, 2011. doi: 10.1016/j.jacc.2011.08.025
4.Freeze SL, Landis BJ, Ware SM, Helm BM: Bicuspid aortic valve: a review with recommendations for genetic counseling.J Genet Couns 25(6):1171–1178, 2016.
正常な胎児循環
胎児循環には以下の特徴がある:
開存している動脈管(肺動脈を大動脈に連結)と卵円孔(右房と左房を連結)を介した血液の右左短絡が換気されていない肺を迂回している
この短絡は,肺細動脈抵抗が大きく,体循環(胎盤循環を含む)の血流への抵抗が比較的小さいことによって促進されている。右心からの拍出量の約90~95%が,肺を迂回して直接体循環へ流れる。胎児の動脈管は,胎児の全身PaO2が低く(約25mmHg),プロスタグランジンの局所産生があることによって開存が維持される。卵円孔は,心房圧の差によって開存が維持される:肺からの血液環流がほとんどないため左房圧は相対的に低いが,胎盤からの大量の血液還流のために右房圧は相対的に高い。
正常な胎児循環
胎児では,心臓の右側に入る血液はすでに胎盤を介して酸素化されている。肺は換気されていないため,肺動脈を通過しなければならない血液は少量である。心臓の右側から出る血液は,ほとんどが以下を介して肺を迂回する:
正常であれば,この2つの構造は出生後まもなく閉鎖する。 |
周産期の変化
最初の数回の呼吸後には,このシステムに著しい変化が生じ,それにより以下の現象が起こる:
肺血流量の増大
卵円孔の機能的閉鎖
肺の拡張,PaO2の上昇,およびPaCO2の低下による血管拡張の結果,肺細動脈抵抗が急激に低下する。肋骨および胸壁の弾性力は肺の間質圧を低下させ,肺毛細血管の血流量をさらに増大させる。肺からの静脈還流量の増大によって左房圧が上昇し,左房と右房との圧力差が減少する;この作用が卵円孔の機能的閉鎖の一因である。
肺循環が確立すると,肺からの静脈還流量が増大して左房圧を上昇させる。空気呼吸はPaO2を増大させ,これにより臍動脈は収縮する。胎盤の血流が減少または途絶し,右房への血液還流を減少させる。こうして右房圧が低下し左房圧が上昇する結果,心房中隔の2つの胎児期構成要素(一次中隔および二次中隔)が共に押され卵円孔を通る血流が止まる。ほとんどの人でこの2つの中隔は最終的に融合し卵円孔は消滅する。
出生後まもなく,胎児の状態から反転して,体血管抵抗が肺血管抵抗より大きい状態へと移行する。これにより,開いている動脈管の血流方向が逆転し,血液の左右短絡が形成される(移行循環と呼ばれる)。この状態は,出生直後(肺血流量の増大および卵円孔の機能的閉鎖が起こる時点)から,動脈管が収縮する約24~72時間後まで持続する。大動脈から動脈管およびその栄養血管へ流入する血液はPO2が高く,このことがプロスタグランジン代謝の変化とともに,動脈管の収縮および閉鎖をもたらす。動脈管が閉鎖すると,成人型循環となる。この時点で,2つの心室は直列ポンプとして働くようになり,肺循環と体循環との間の大きな短絡は存在しない。
出生直後の数日間,ストレスを受けた新生児では胎児循環に戻ることがある。低酸素症および高炭酸ガス血症を伴う仮死は,肺細動脈の収縮および動脈管の拡張を引き起こし,上述の過程を逆行させ,今や開いている動脈管,再開通した卵円孔,またはその両方を介した右左短絡を生じさせる。その結果,このような新生児は,遷延性肺高血圧症または胎児循環遺残(臍帯循環は存在しないが)と呼ばれる重度の低酸素状態に陥る。治療の目標は,肺血管収縮の原因となった状態を元に戻すことである。
病態生理
先天性心奇形は次のように分類される(先天性心奇形の分類の表を参照):
チアノーゼ性
非チアノーゼ性(左右短絡または閉塞性病変)
先天性心奇形の生理学的な影響は,無症状の小児でみられる心雑音または脈拍異常から,重度のチアノーゼ,心不全,循環虚脱まで,非常に多彩である。
チアノーゼ性心奇形
程度は様々であるが,脱酸素化された静脈血が左心に短絡すると(右左短絡),体循環系の動脈血酸素飽和度が低下する。
デオキシヘモグロビン濃度が5g/dL(50g/L)を超えると,チアノーゼが出現する。チアノーゼの持続による合併症としては,赤血球増多,ばち指,血栓塞栓症(脳卒中など),出血性疾患,脳膿瘍,高尿酸血症などがある。未修復のファロー四徴症の乳児では,高度チアノーゼ発作(hypercyanotic spell)が起こりうる。
肺血流量は奇形の種類に応じて減少,正常,または増加(しばしばチアノーゼに加えて心不全を引き起こす)となり,結果として様々な重症度のチアノーゼを呈する。心雑音が様々な大きさで聴取されるが,特異的ではない。
左右短絡
左心(左房または左室)または大動脈から供給される酸素化された血液が,左心と右心をつなぐ開口部または交通を通って,右心(右房または右室)または肺動脈へ短絡する。
出生直後は肺血管抵抗が高いため,この交通を通る血流は軽微であるか,両方向性である。しかしながら,生後24~48時間以内には,肺血管抵抗の低下が進み,左心系から右心系へ流れる血流量が増加する。右側に流れ込んだ余分な血流は,肺血流量と肺動脈圧を様々な程度まで増大させる。この増大幅が大きいほど,より重度の症状が生じ,少量の左右短絡では典型的には症状や徴候は生じない。
高圧の短絡(心室または大血管レベルで生じるもの)は生後数日ないし数週間後に明らかになるが,低圧の短絡(心房中隔欠損)はかなり後になってから明らかとなる。無治療の場合,肺血流量の増加と肺動脈圧の上昇によって肺血管疾患が引き起こされ,最終的にはアイゼンメンジャー症候群に至る。大量の左右短絡(例,大きな心室中隔欠損症[VSD],動脈管開存症[PDA])では,過度の肺血流および左室容量負荷が生じることで,心不全の徴候が出現し,乳児期にはしばしば発育不良を来す。大量の左右短絡は,肺コンプライアンスの低下と気道抵抗の上昇にもつながる。これらの因子は,RSウイルス感染症やその他の上気道または下気道感染症のある乳児において入院のリスクを高める。
閉塞性病変
心不全
先天性心奇形の中には,血行動態に有意な変化を起こさないものも存在する(例,大動脈二尖弁,軽度の大動脈弁狭窄)。それ以外の心奇形では,圧負荷または容量負荷が生じる結果,ときに心不全を来す。心不全は,心拍出量が身体の代謝要求を満たすのに不十分となった場合,または心臓が静脈還流量を十分に処理できなくなった場合に発生し,肺うっ血(左室不全),各体位で下方にある組織および腹部内臓で主に生じる浮腫(右室不全),またはその両方を引き起こす。乳児期および小児期における心不全の原因としては,先天性心奇形以外にも多くのものが存在する(小児における心不全の一般的な原因の表を参照)。
動脈管依存性先天性心疾患
症状と徴候
先天性心疾患の臨床像は多様であるが,一般的には以下のものがみられる:
雑音
チアノーゼ
心不全
脈拍減弱または脈拍欠損
このほかに身体診察でみられる異常としては,循環性ショック,灌流不良,異常なII音(S2―単一または大幅な分裂),収縮期クリック,奔馬調律,徐脈,頻脈,不整脈などがある。
雑音
左右短絡および閉塞性病変の大半では収縮期雑音が発生する。収縮期雑音および振戦(スリル)は,発生源に最も近い体表部位で最も著明となるため,その位置の同定は診断に役立つ。肺動脈弁または大動脈弁を通過する血流量が増加すると,漸増漸減性の収縮中期(駆出性収縮期)雑音が発生する。房室弁を介した逆流または心室中隔欠損を介した血流が存在する場合には,全収縮期(汎収縮期)雑音が発生し,その強度が上がるにつれてI音(S1)が不明瞭になる。
動脈管開存症では,収縮期と拡張期を通して動脈管に血液が流れるため,II音(S2)で中断しない連続性雑音が発生するのが典型的である。この雑音は2つの音調から成り,収縮期の音(より高い圧により生じる)は拡張期の音よりはっきり聞こえる。
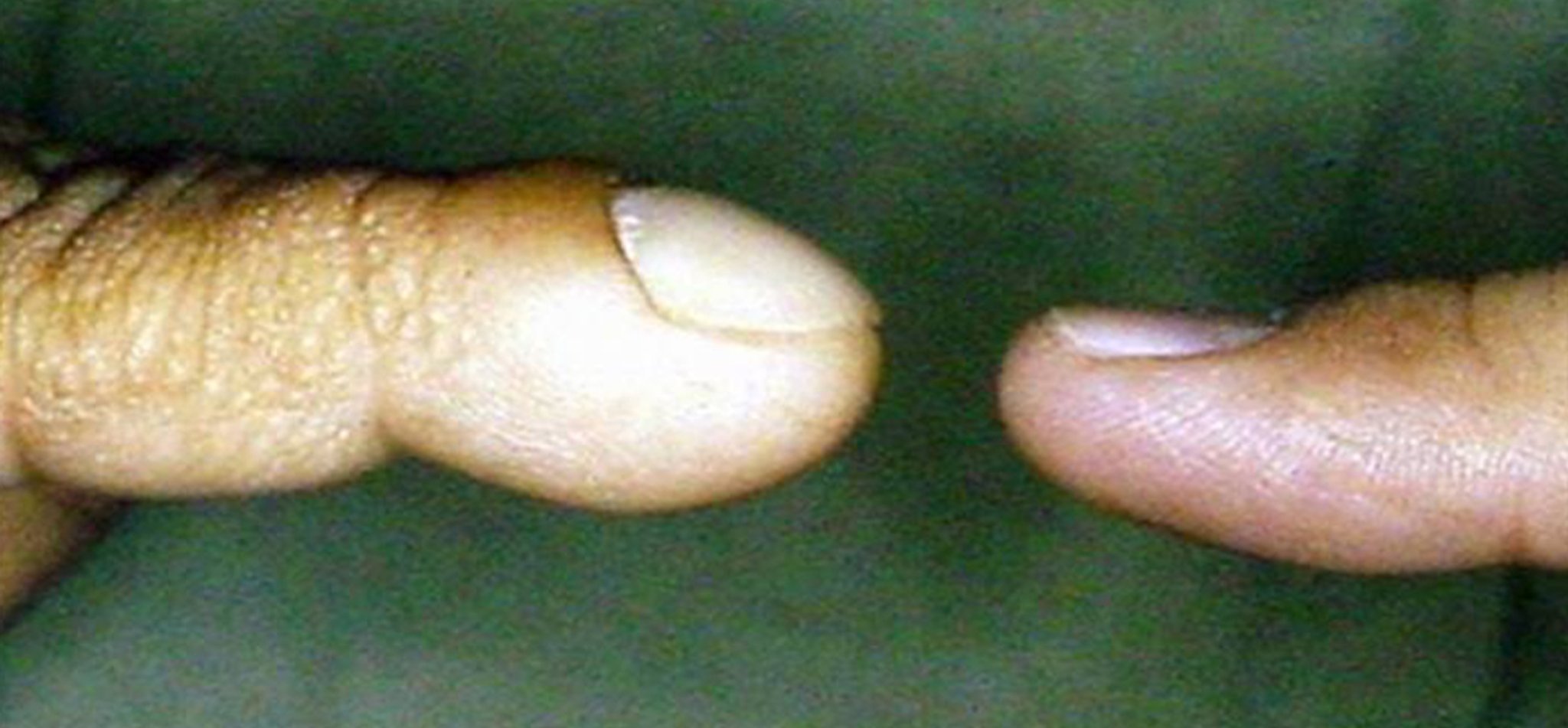
チアノーゼ

© Springer Science+Business Media
中枢性チアノーゼは,口唇と舌および/または爪床の青色調変色を特徴とし,これはデオキシヘモグロビンが増加している(5g/dL[50g/L]以上)ときにみられ,血中酸素レベルの低下(通常は酸素飽和度85%未満)を意味する。口唇または爪床チアノーゼを伴わない口囲および肢端チアノーゼ(手足のチアノーゼ)は,低酸素血症ではなく末梢血管の収縮により起こるものであり,新生児では一般的な正常所見である。チアノーゼが長期間持続する児童では,ばち指がみられることが多い。
心不全
乳児では,心不全の症状または徴候として以下のものがみられる:
頻脈
頻呼吸
哺乳中の呼吸困難
発汗,特に哺乳中
不穏,易刺激性
肝腫大
哺乳時に呼吸困難が起こると,哺乳量が不十分になって発育不良を呈するが,これは心不全による代謝必要量の増加や頻回の気道感染症によって,さらに悪化する場合がある。成人や児童とは対照的に,ほとんどの乳児では頸静脈怒張や就下性の浮腫を呈することはないが,ときに眼窩周囲に浮腫が発生する。肝腫大は乳児の心不全における特徴的な所見である。心不全の児童でみられる所見は,成人患者のそれと同様である。
心奇形のその他の臨床像
新生児期には,一部の心奇形(例,左心低形成症候群,重篤な大動脈弁狭窄,大動脈弓離断,大動脈縮窄症)において循環性ショックが最初の臨床像となることがある。粘膜の蒼白化またはチアノーゼがみられ,四肢は冷たく,脈は微弱になり,血圧は低く,刺激に対する反応も減弱するなど,極めて病的な様相を呈する。
小児にみられる胸痛は,通常は心臓性ではない。乳児では,胸痛は原因不明の著明な易刺激性(特に哺乳中または哺乳後)として顕在化し,肺動脈からの左冠動脈起始異常によって生じる可能性がある。児童および青年では,心臓の病態に起因する胸痛は労作時に認められるのが通常で,冠動脈異常,心膜炎,心筋炎,または高度の大動脈弁狭窄によって生じる可能性がある。
失神は典型例では前兆症状なしに,しばしば労作に関連して生じるが,心筋症(肥大型または拡張型),冠動脈起始異常,または遺伝性不整脈症候群(例,QT延長症候群,カテコールアミン誘発性多形性心室頻拍[CPVT],ブルガダ症候群)など特定の異常に伴って生じる可能性がある。高校生年代のアスリートで最もよくみられる。
診断
パルスオキシメトリーによるスクリーニング
心臓の診察
心電図および胸部X線検査
心エコー検査
ときに,心臓MRIまたはCT血管造影,心臓カテーテル検査と心血管造影
心雑音,チアノーゼ,脈拍異常,または心不全症状がみられる場合は,先天性心疾患が示唆される。そのような新生児では,心エコー検査を行って先天性心疾患の診断を確定する。チアノーゼが唯一の異常である場合は,メトヘモグロビン血症を除外すべきである。
典型的には心エコー検査で診断可能であるが,心臓MRIまたはCT血管造影で,重要な解剖学的詳細を明らかにできる場合がある。奇形の確定診断または重症度の術前評価のため,ときに心臓カテーテル検査と心血管造影が必要になることがあり,治療を目的として行うことも増えてきている。
新生児スクリーニング
新生児期の先天性心疾患は臨床像が軽微となる場合や全く症候がみられない場合がある一方,重症先天性心疾患(critical congenital heart disease)の検出の失敗や遅れは新生児死亡や重大な合併症の発生につながる可能性があり,特に生後数時間または数日中に外科的治療や入院下での内科的治療を必要とする新生児の10~15%では,その可能性が高くなる。そのため,退院前に新生児全例に対してパルスオキシメトリーによる重症先天性心疾患のスクリーニングを実施することが推奨されている。生後24時間以上の時点でもスクリーニングを行い,以下のいずれかに1つでも該当する場合は陽性と判定する:
部位を問わず酸素飽和度が90%未満である。
右手と右足での酸素飽和度が,1時間間隔で実施した3回の測定においてともに95%未満である。
右手(動脈管前)と右足(動脈管後)での酸素飽和度の絶対差が,1時間間隔でそれぞれ行った3回の測定で3%を超える。
スクリーニングで陽性となった新生児には,先天性心疾患および低酸素血症の他の原因(例,様々な呼吸器疾患,中枢神経系の抑制,敗血症)に関する包括的評価を全例で行うべきであり,典型的には胸部X線,心電図,心エコー検査のほか,しばしば血液検査などを施行する。パルスオキシメトリーによるスクリーニングの感度は75%を若干超える程度で,最も見逃されやすい先天性心疾患の病変は左室の閉塞性病変(例,大動脈縮窄)である。
治療
内科的治療による心不全の安定化(例,利尿薬,アンジオテンシン変換酵素(ACE)阻害薬,β遮断薬,ジゴキシン,スピロノラクトン,塩分制限,選択された症例では酸素またはプロスタグランジンE1の投与)
外科的修復またはカテーテルインターベンション
心不全の治療は病因によって大きく異なる。根治的治療としては,典型的には基礎にある問題の是正が必要である。
急性心不全症状またはチアノーゼを内科的治療で安定させた後には,ほとんどの患児で外科的またはカテーテル手技による修復が必要となるが,経時的に縮小ないし閉鎖する可能性が高い特定の心室中隔欠損症と軽度の弁機能障害は例外である。カテーテル治療としては以下のものがある:
重度のチアノーゼを呈する大血管転位症の新生児に対する症状緩和を目的としたバルーン心房中隔裂開術
新生児における心不全
出生後最初の1週間に生じた重度の急性心不全またはチアノーゼは,医学的緊急事態である。確実な血管アクセスを確保すべきであり,臍静脈カテーテルによるものが望ましい。
重症先天性心疾患が疑われるか確定診断されている場合は,プロスタグランジンE1の点滴静注を0.05~0.1μg/kg/分から開始すべきである。この時期に発症する心疾患の大半では,体血流(例,左心低形成症候群,重篤な大動脈弁狭窄症,大動脈縮窄症)または肺血流(危機的な肺動脈弁狭窄症,肺動脈閉鎖症,重症ファロー四徴症などのチアノーゼ性心疾患)のいずれかを動脈管に依存していることから,動脈管の開存を維持することが重要となる。
重症(critically ill)の新生児では機械的人工換気がしばしば必要になる。酸素投与は肺血管抵抗を下げることになるが,これは特定の異常(例,左心低形成症候群)を有する乳児には有害となるため,酸素投与は慎重に行うか,場合によっては控えるべきである。
新生児心不全に対するその他の治療としては,利尿薬,強心薬,後負荷を軽減する薬剤などがある。利尿薬のフロセミドは,まず1mg/kgを急速静注し,尿量に基づいて漸増する。強心薬のドパミンまたはドブタミンは,点滴により血圧維持を補助できるが,心拍数と後負荷を増加させることで心筋酸素消費量が増大するという短所もある。これらの薬剤が先天性心疾患の乳児で使用されることはまれである。先天性心疾患の術後患者に頻用されるミルリノンは,陽性変力作用と血管拡張作用の両方を有している。ドパミン,ドブタミン,ミルリノンは全て不整脈リスクを増大させる可能性がある。純粋な血管拡張薬であるニトロプルシドは,術後高血圧に使用されることがある。0.3~0.5μg/kg/分で開始し,期待する効果が得られるまで漸増する(通常の維持量は約3μg/kg/分)。
月齢の高い乳児および小児における心不全
治療法としては,しばしば利尿薬(例,フロセミド0.5~1mg/kg,静注または1~3mg/kg,経口,8~24時間毎,必要に応じて漸増)やACE阻害薬(例,カプトプリル0.1~0.3mg/kg,経口,1日3回)が用いられる。カリウム保持性利尿薬(例,スピロノラクトン1mg/kg,経口,1日1回または1日2回,必要に応じて2mg/kg/回まで漸増)も有用となりうる(特に高用量のフロセミドが必要な場合)。β遮断薬(例,カルベジロール,メトプロロール)は慢性うっ血性心不全の患児にしばしば追加される。
ジゴキシンは以前より使用されなくなったが,大量の左右短絡がある心不全患者,特定の先天性心疾患術後患者では,なお有用となりうる(用量は年齢により異なる;小児におけるジゴキシンの経口用量の表を参照)。注目すべきことに,ジゴキシンは,単心室症に対するNorwood手術後から第2期手術までの間に使用することで,死亡率を低下させる(1)。新生児上室頻拍の治療において,第1選択薬としてのジゴキシンの使用は,プロプラノロールよりも死亡率が高くなることから,使用される機会が減少している(2)。しかしながら,もしWolff-Parkinson-White(WPW)症候群がなければ,プロプラノロールが無効に終わった場合の第一の薬剤として,またはプロプラノロールやその他の抗不整脈薬と併用する第二の薬剤として,ジゴキシンが有用となる可能性がある。
酸素投与は心不全における低酸素血症を緩和して呼吸窮迫を軽減する可能性があるが,肺上皮損傷リスクを最小限にするため,可能であれば,吸入気酸素分画(FIO2)を< 40%に維持すべきである。酸素投与は,肺循環を増大させるため,左右短絡病変または左心系閉塞性疾患の患者では(使用する場合は)注意して使用すべきである。
一般に,食塩制限を含めた健康的な食事が推奨されるが,具体的な疾患や臨床像に応じて食習慣の変更が必要になることもある。心不全が代謝必要量を増加させる一方,合併する呼吸困難が哺乳をさらに困難にする。重症先天性心疾患を有する乳児では,特に左心系に閉塞性病変がある場合,壊死性腸炎のリスクを最小限に抑えるために授乳を控えることもある。左右短絡病変により心不全を来している乳児では,カロリー量を増やした栄養が推奨され,それによりカロリー補給量を増やすとともに,容量負荷のリスクを低減する。成長を維持するために経管栄養が必要になる場合もある。これらの対策で体重増加が得られない場合は,奇形の外科的修復が適応となる。
心内膜炎予防
心内膜炎予防に関するAmerican Heart Associationの最新ガイドラインでは,以下に該当する先天性心疾患を有する患児には抗菌薬の予防投与が必要とされている:
未修復のチアノーゼ性先天性心疾患(姑息的な短絡および導管手術を受けた患児を含む)
完全に修復された先天性心疾患で,人工材料またはデバイスが使用された場合は術後6カ月間
修復術後の先天性心疾患のうち,人工パッチまたはデバイスの使用部位またはその隣接部に欠損が遺残しているもの
機械弁または生体弁
心内膜炎の既往
治療に関する参考文献
1.Oster ME, Kelleman M, McCracken C, et al: Association of digoxin with interstage mortality: Results from the Pediatric Heart Network Single Ventricle Reconstruction Trial Public Use Dataset.J Am Heart Assoc 5(1): e002566., 2016.
2.Bolin EH, Lang SM, Tang X, et al: Propranolol versus digoxin in the neonate for supraventricular tachycardia (from the Pediatric Health Information System).Am J Cardiol 119(10): 1605–1610, 2017.
より詳細な情報
以下の英語の資料が有用であろう。ただし,本マニュアルはこれらの資料の内容について責任を負わないことに留意されたい。
American Heart Association: Common Heart Defects: Provides overview of common congenital heart defects for parents and caregivers
American Heart Association: Infective Endocarditis: Provides an overview of infective endocarditis, including summarizing prophylactic antibiotic use, for patients and caregivers







