


Retinoblastoma is a cancer of the retina, the light-sensing area at the back of the eye.
Retinoblastomas can result from a genetic mutation.
The child may have a white pupil or cross-eyes or occasionally vision problems.
Doctors can often diagnose retinoblastoma by looking into the eye with a special instrument while the child is under anesthesia.
Treatment may involve surgery, chemotherapy, or sometimes radiation therapy.
(See also Overview of Childhood Cancer.)
Retinoblastomas represent about 2% of childhood cancers and almost always occur in children under 2 years of age. Retinoblastomas occur only in children.
This cancer results from a mutation in certain genes that control eye development. Sometimes, the mutation is passed on (inherited) from a parent. At other times, it occurs spontaneously (not inherited) very early during development of the embryo.
When the mutation is inherited, affected children may pass the mutation on to their children. There is a 50% chance that the mutation will be passed on if one parent has the mutation. If the mutation is passed on, most children with the mutation will develop retinoblastoma. Retinoblastoma is hereditary in all children with cancer in both eyes and in 15% of children with cancer in one eye.
At other times, the mutation does not occur until later in embryonic development and only in the embryo’s eye cells. In such cases, the mutation is not inherited and cannot be passed on to children.
Viewing the Retina

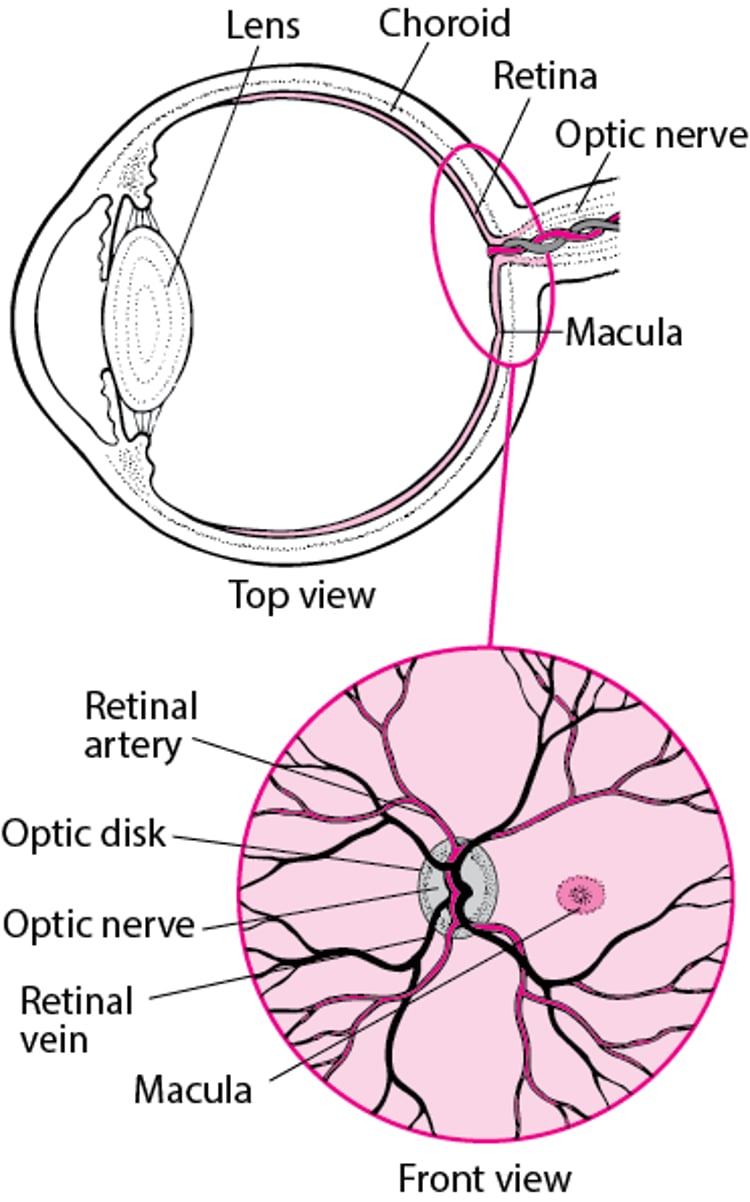
Retinoblastoma does not usually spread beyond the eye, but it occasionally spreads to the brain along the optic nerve (the nerve that leads from the eye to the brain). It may rarely spread to other locations, such as the bone marrow and bones.
Symptoms of Retinoblastoma
Symptoms of retinoblastoma can include a white pupil (leukocoria) or cross-eyes (strabismus).

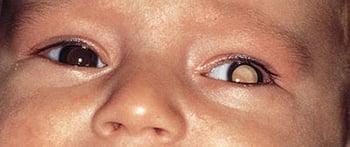

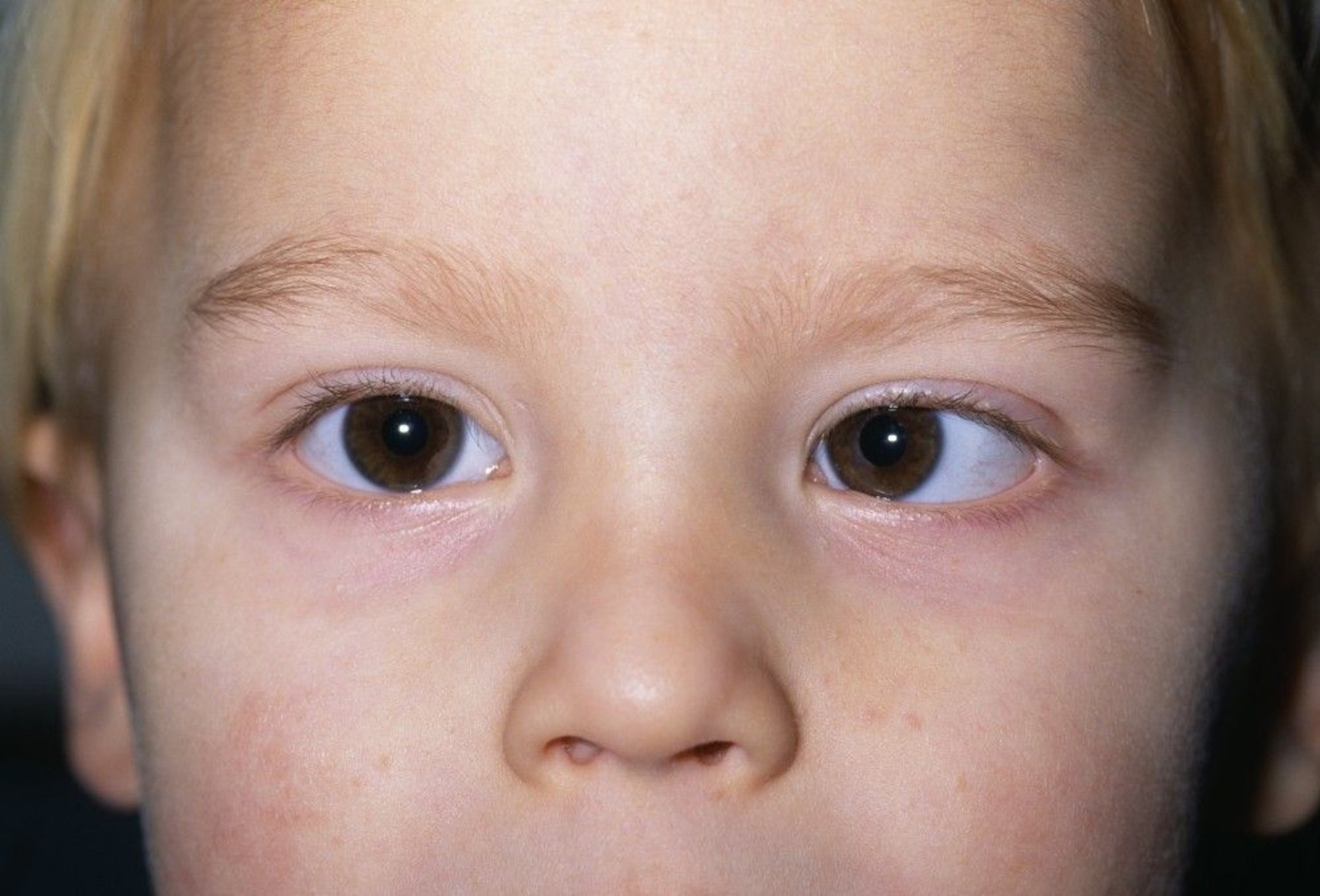
DR P. MARAZZI/SCIENCE PHOTO LIBRARY
Large retinoblastomas may affect vision but tend to cause few other symptoms.
If the cancer has spread, symptoms may include headache, loss of appetite, or vomiting.
Diagnosis of Retinoblastoma
Looking into the eye with a special instrument while the child is under anesthesia
Ultrasonography, computed tomography (CT), or magnetic resonance imaging (MRI) and sometimes optical coherence tomography
Sometimes a bone scan, examination of bone marrow, and spinal tap
If a doctor suspects a retinoblastoma, the child is given a general anesthetic, which makes the child lose consciousness, and both eyes are examined. A light and a special lens (indirect ophthalmoscopy) are used to look through the lens and iris at the retina. A general anesthetic is necessary because small children are not able to cooperate during the careful, time-consuming examination required to diagnose retinoblastoma.
The cancer can also be identified by ultrasonography, CT, or MRI of the eyes. These tests also help determine whether the cancer has spread to the brain. Optical coherence tomography is another imaging test that sometimes is done.
Doctors may also do a spinal tap (lumbar puncture) to look for cancer cells in a sample of cerebrospinal fluid. Finding cancer cells in this fluid is further evidence that the cancer has spread to the brain.
Because the cancer can spread to the bones or bone marrow, a bone scan may be done and a sample of bone marrow may be removed for examination.
Children who have retinoblastoma should see a genetic specialist and have genetic testing. The specialist can then advise parents whether other family members are at risk and whether any other tests should be done. Typically, if children have a hereditary retinoblastoma gene, their parents and brothers and sisters should also be tested for the mutated gene. Siblings who have the mutated gene should have their eyes examined for retinoblastoma beginning at birth and continuing every 4 months until they are 4 years old.
If genetic testing is not available, all children who have a parent or sibling who had retinoblastoma should have the same regular eye examinations. Adult family members of a child with retinoblastoma also need to have an eye examination. Even though adults will not develop retinoblastoma, the gene that causes retinoblastoma can also cause a noncancerous (benign) eye tumor called retinocytoma.
Treatment of Retinoblastoma
Surgical removal of the eye
Chemotherapy
Radiation therapy, lasers, and cryotherapy
(See also Cancer Treatment Principles and Surgery for Cancer.)
When only one eye is affected and that eye has little or no vision, doctors usually remove the entire eyeball along with part of the optic nerve.
When the cancer affects both eyes, doctors try to preserve some vision by treating the cancer without removing both eyeballs, although they sometimes remove the most severely affected eye. Treatment options include chemotherapy drugs injected directly through the main artery that provides blood to the eye (called intra-arterial chemotherapy), radiation therapy, lasers, freezing (cryotherapy), or, for very small cancers, patches containing radioactive material (brachytherapy).
Combinations of chemotherapy drugs
Radiation therapy to the eye has serious consequences, such as cataracts, decreased vision, chronic dry eye, and wasting of the tissue around the eye. The bones of the face may not grow normally, resulting in a deformed appearance. Additionally, the risk of a developing a second cancer increases in the area where radiation is done.
After treatment, a doctor who specializes in treating cancer in children (pediatric oncologist) and an ophthalmologist should continue to monitor the child because of the risk of a second cancer developing.
Prognosis for Retinoblastoma
With treatment, children with retinoblastoma that has not spread past the retina are cured more than 90% of the time. The prognosis is poor for children whose cancer has spread.
Without treatment, most children with retinoblastoma die within 2 years.
Children with the hereditary type of retinoblastoma have an increased risk of developing a second cancer, such as soft-tissue sarcomas, melanomas, and osteosarcomas. About half of second cancers occur where radiation therapy was given. About 70% of the time, the second cancer occurs within 30 years of the retinoblastoma.
More Information
The following English-language resource may be useful. Please note that THE MANUAL is not responsible for the content of this resource.
American Cancer Society: If Your Child Is Diagnosed With Cancer: A resource for parents and loved ones of a child who has cancer that provides information about how to cope with some of the problems and questions that come up just after a child is diagnosed






