


Eosinophilic granulomatosis with polyangiitis is a systemic small- and medium-vessel necrotizing vasculitis, characterized by extravascular granulomas, eosinophilia, and tissue infiltration by eosinophils. It occurs in people with adult-onset asthma, allergic rhinitis, nasal polyposis, or a combination. Diagnosis is best confirmed by biopsy. Treatment is primarily with corticosteroids plus another immunosuppressant.
(See also Overview of Vasculitis.)
Eosinophilic granulomatosis with polyangiitis (EGPA) occurs in about 3 people/million. Mean age at onset is 48.
EGPA is characterized by extravascular necrotizing granulomas (usually rich in eosinophils), eosinophilia, and tissue infiltration by eosinophils. However, these abnormalities do not always coexist. The vasculitis typically affects small- and medium-sized arteries. Any organ can be affected, but the lungs, skin, sinuses, cardiovascular system, kidneys, peripheral nervous system, central nervous system, joints, and gastrointestinal (GI) tract are most commonly affected. Rarely, pulmonary capillaritis may cause alveolar hemorrhage.
Etiology of EGPA
The cause of eosinophilic granulomatosis with polyangiitis is unknown. However, an allergic mechanism, with tissue directly injured by eosinophils and neutrophil degranulation products, may be involved. Activation of T lymphocytes seems to help maintain eosinophilic inflammation. The syndrome occurs in patients who have adult-onset asthma, allergic rhinitis, nasal polyposis, or a combination. Antineutrophil cytoplasmic autoantibodies (ANCA) are present in 30% to 40% of cases.
Symptoms and Signs of EGPA
The syndrome has 3 phases, which may overlap:
Prodromal: This phase may persist for years. Patients have allergic rhinitis, nasal polyposis, asthma, or a combination.
2nd phase: Peripheral blood and tissue eosinophilia is typical. Clinical presentation, which may resemble Löffler syndrome, includes chronic eosinophilic pneumonia and eosinophilic gastroenteritis.
3rd phase: Potentially life-threatening vasculitis develops. Organ dysfunction and systemic symptoms (eg, fever, malaise, weight loss, fatigue) are common in this phase.
However, the phases do not necessarily follow one another consecutively, and the time interval between them varies greatly.
Various organs and systems may be affected:
Respiratory: Asthma, often with onset during adulthood, occurs in most patients and tends to be severe and corticosteroid-dependent. Sinusitis is common, but not destructive, without severe necrotizing inflammation. Patients may be short of breath. Transient patchy pulmonary infiltrates are common.
Neurologic: Neurologic manifestations are very common. Multiple mononeuropathy (mononeuritis multiplex) occurs in up to three fourths of patients. Central nervous system involvement is rare but can include hemiparesis, confusion, seizures, and coma, with or without cranial nerve palsies or evidence of cerebral infarction.
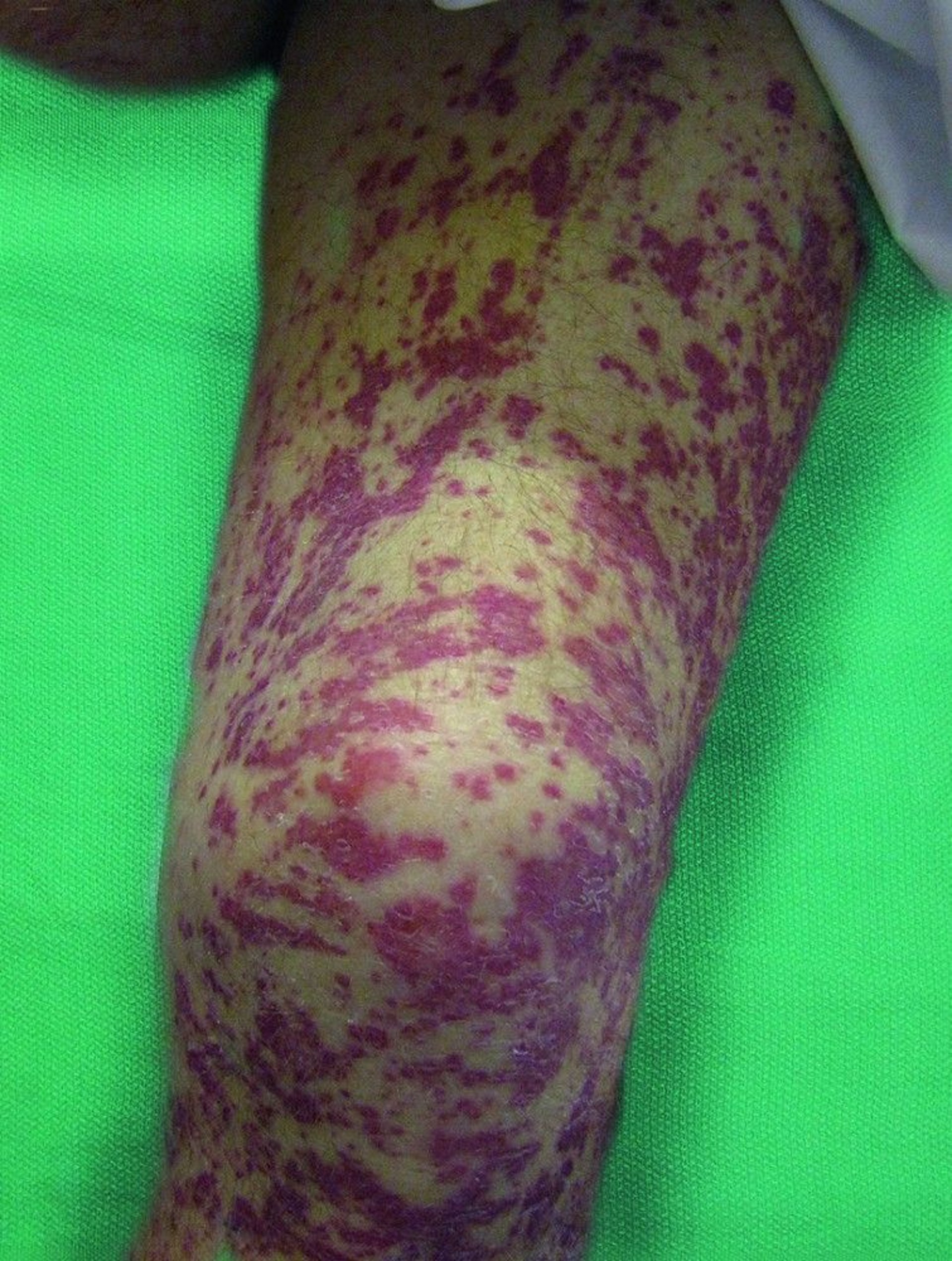
Cutaneous: The skin is affected in about one half of patients. Nodules and papules appear on extensor surfaces of extremities. They are caused by extravascular palisading granulomatous lesions with central necrosis. Purpura or erythematous papules, due to leukocytoclastic vasculitis with or without prominent eosinophilic infiltration, may develop.
Musculoskeletal: Arthralgias, myalgias, or even arthritis can occur.
Cardiac: Cardiac involvement, a major cause of mortality, includes heart failure due to myocarditis and endomyocardial fibrosis, coronary artery vasculitis (possibly with myocardial infarction), valvular disorders, and pericarditis. The predominant histopathologic finding is eosinophilic myocarditis.
Gastrointestinal: Up to one third of patients present with gastrointestinal symptoms (eg, abdominal pain, diarrhea, bleeding, acalculous cholecystitis) due to eosinophilic gastroenteritis or mesenteric ischemia due to vasculitis.
Renal: The kidneys are affected less often than in other vasculitic disorders associated with antineutrophil cytoplasmic autoantibodies. Typically, pauci-immune (few if any immune complexes), focal segmental necrotizing glomerulonephritis with crescent formation is present; eosinophilic or granulomatous inflammation of the kidneys is rare.

© Springer Science+Business Media
Renal, cardiac, or neurologic involvement indicates a worse prognosis.
Diagnosis of EGPA
Clinical criteria
Routine laboratory tests
Echocardiography
Biopsy
The 2012 Chapel Hill Consensus Conference (1) defined eosinophilic granulomatosis with polyangiitis (EGPA) as an eosinophil-rich and necrotizing granulomatous inflammation involving the respiratory tract with necrotizing vasculitis of small- and medium-sized vessels in association with asthma and eosinophilia. Criteria for classification from the American College of Rheumatology consist of the following:
Asthma
Eosinophilia of > 10% in peripheral blood
Paranasal sinusitis
Pulmonary infiltrates, sometimes transient
Histologic evidence of vasculitis with extravascular eosinophils
Multiple mononeuropathy or polyneuropathy
If ≥ 4 criteria are present, sensitivity is 85%, and specificity is 99.7%.
Testing aims to establish the diagnosis and the extent of organ involvement and to distinguish EGPA from other eosinophilic disorders (eg, parasitic infections, drug reactions, acute eosinophilic pneumonia and chronic eosinophilic pneumonia, allergic bronchopulmonary aspergillosis, hypereosinophilic syndrome). Diagnosis of EGPA is suggested by clinical findings and results of routine laboratory tests but should usually be confirmed by biopsy of lung or other affected tissue.
Blood tests and chest x-rays are done, but results are not diagnostic. Complete blood count with differential is done to check for eosinophilia, which in some patients may be a marker of disease activity. IgE and C-reactive protein levels and erythrocyte sedimentation rate (ESR) are determined periodically to evaluate inflammatory activity. Urinalysis and creatinine are done to screen for renal disease and monitor its severity. Electrolyte levels are measured.
Serologic testing is done and detects antineutrophil cytoplasmic autoantibodies (ANCA) in up to 40% of patients; if ANCA is detected, enzyme-linked immunosorbent assay (ELISA) is done to check for specific antibodies. Perinuclear ANCA (p-ANCA) with antibodies against myeloperoxidase is the most common result, but ANCA is not a specific or sensitive test for EGPA.
Although used as markers of disease activity, eosinophilia, IgE, ANCA, ESR, and C-reactive protein levels accomplish this and predict flare-ups only with significant limitations.
Chest x-ray often shows transient patchy pulmonary infiltrates.
A 2D echocardiogram of the heart should be obtained in all patients at baseline and repeated over time if symptoms and/or signs of heart failure develop.
Biopsy of the most accessible affected tissue should be done if possible.
Diagnosis reference
1. Jennette JC, Falk RJ, Bacon PA, et al: 2012 Revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum 65(1):1-11, 2013. doi: 10.1002/art.37715
Treatment of EGPA
Corticosteroids
granulomatosis with polyangiitis or microscopic polyangiitis1
Cardiac involvement as manifested by myocarditis with heart failure is the main cause of death in EGPA.
Treatment reference
Wechsler ME, Akuthota P, Jayne D, et al: Mepolizumab or placebo for eosinophilic granulomatosis with polyangiitis. N Engl J Med 376(20):1921-1932, 2017. doi:10.1056/NEJMoa1702079
Key Points
Eosinophilic granulomatosis with polyangiitis is a rare small- and medium-sized vessel vasculitis.
Phases include upper respiratory symptoms and wheezing, eosinophilic pneumonia and gastroenteritis, and life-threatening vasculitis.
Phases may occur in or out of order and may overlap.
Cardiac or neurologic involvement can occur and indicate a poor prognosis.
Diagnose by clinical criteria, routine laboratory testing, and sometimes biopsy.
Do a 2D echocardiogram in all patients.
Treat with corticosteroids and other immunosuppressants, based on severity of disease, and use the same criteria for treatment of granulomatosis with polyangiitis or microscopic polyangiitis.
More Information
The following English-language resource may be useful. Please note that THE MANUAL is not responsible for the content of this resource.
American College of Rheumatology (ACR): Lists a number of ACR-approved classification criteria sets and provides information about the purpose of criteria sets, their development and validation, and the role of the ACR in adopting them.







