


Bệnh mô liên kết hỗn hợp ít gặp, là một hội chứng đặc trưng bởi các triệu chứng lâm sàng của các bệnh lupus ban đỏ hệ thống, xơ cứng vì toàn thể và viêm đa cơ với nồng độ rất cao của kháng thể kháng nhân kháng lại kháng nguyên ribonucleoprotein. Sưng bàn tay, hội chứng Raynaud, đau nhiều khớp, viêm cơ, giảm nhu động thực quản, viêm phổi kẽ là các tổn thương hay gặp. Chẩn đoán xác định dựa trên kết hợp các triệu chứng lâm sàng, kết quả xét nghiệm kháng thể kháng ribonucleoprotein dương tính, và các kháng thể đặc hiệu cho các bệnh tự miễn khác âm tính. Điều trị khác nhau tùy theo mức độ nghiêm trọng của bệnh và sự tham gia của cơ quan nhưng thường bao gồm corticosteroid và các thuốc ức chế miễn dịch khác.
Bệnh mô liên kết hỗn hợp (MCTD) gặp ở các chủng tộc trên toàn thế giới, tỷ lệ cao nhất ở thanh thiếu niên và những người trong độ tuổi 20. Khoảng 80% người mắc bệnh là phụ nữ. Nguyên nhân của MCTD chưa rõ. Ở nhiều bệnh nhân, bệnh phát triển thành xơ cứng bì hệ thống hoặc là lupus ban đỏ hệ thống (SLE).
Triệu chứng và dấu hiệu của MCTD
Hội chứng Raynaud (Hiện tượng Raynaud) có thể xuất hiện trước những biểu hiện khác nhiều năm. Thông thường, các biểu hiện đầu tiên giống như SLE sớm, xơ cứng hệ thống, viêm đa cơ, hoặc thậm chí là viêm khớp dạng thấp. Nhiều bệnh nhân khởi đầu có biểu hiện của bệnh mô liên kết không xác định. Các biểu hiện của bệnh có thể tiến triển nặng lên và tổn thương ở nhiều cơ quan, và triệu chứng lâm sàng thay đổi theo thời gian.
ban đầu, sưng lan tỏa các khớp bàn tay điển hình nhưng không phổ biến. Các dạng tổn thương da giống như bệnh lupus hay viêm da cơ. Tổn thương da giống như xơ cứng hệ thống lan tỏa thay đổi và có thể có hoại tử do thiếu máu hoặc loét ở đầu ngón tay.

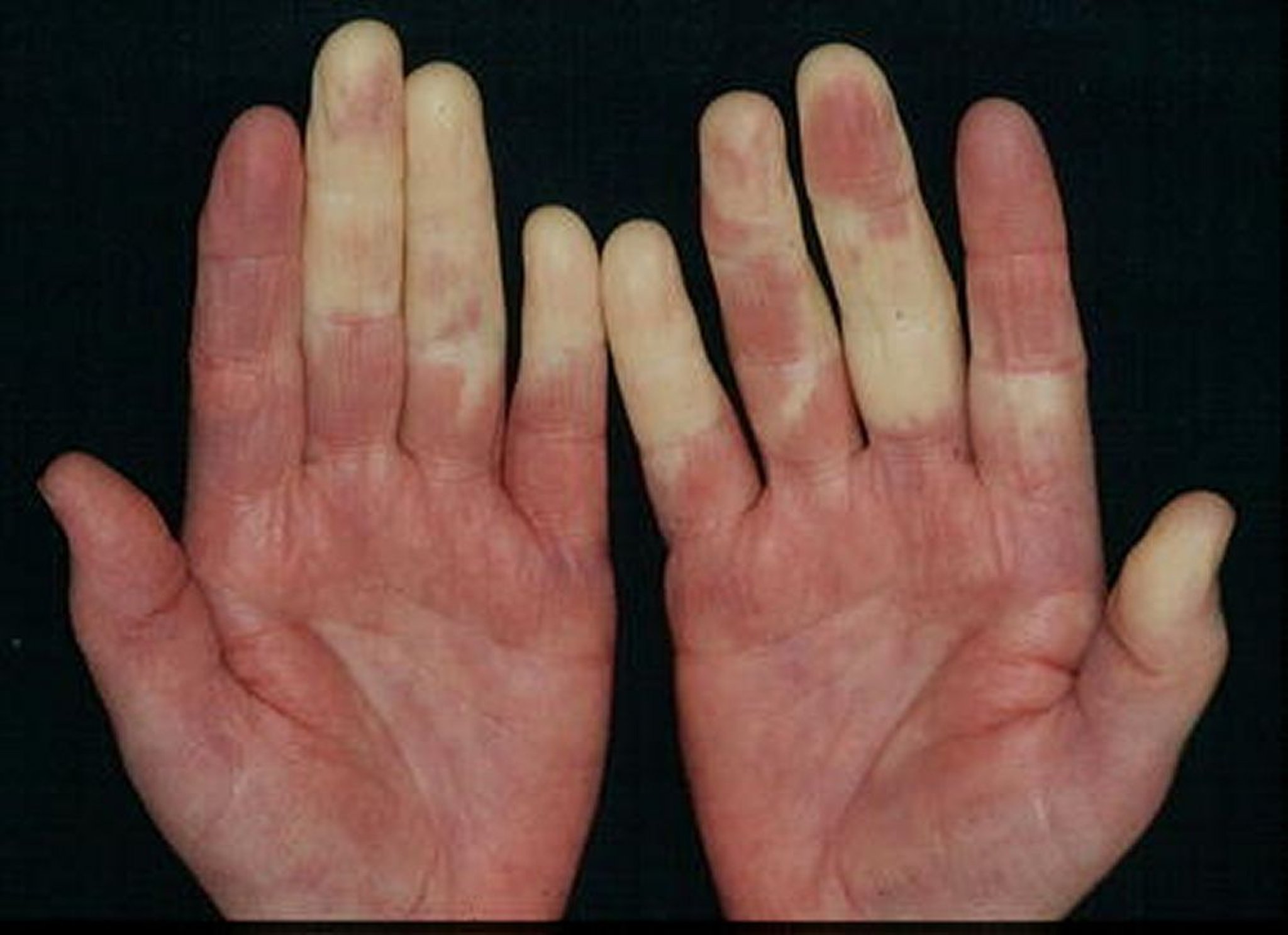
© Springer Science+Business Media

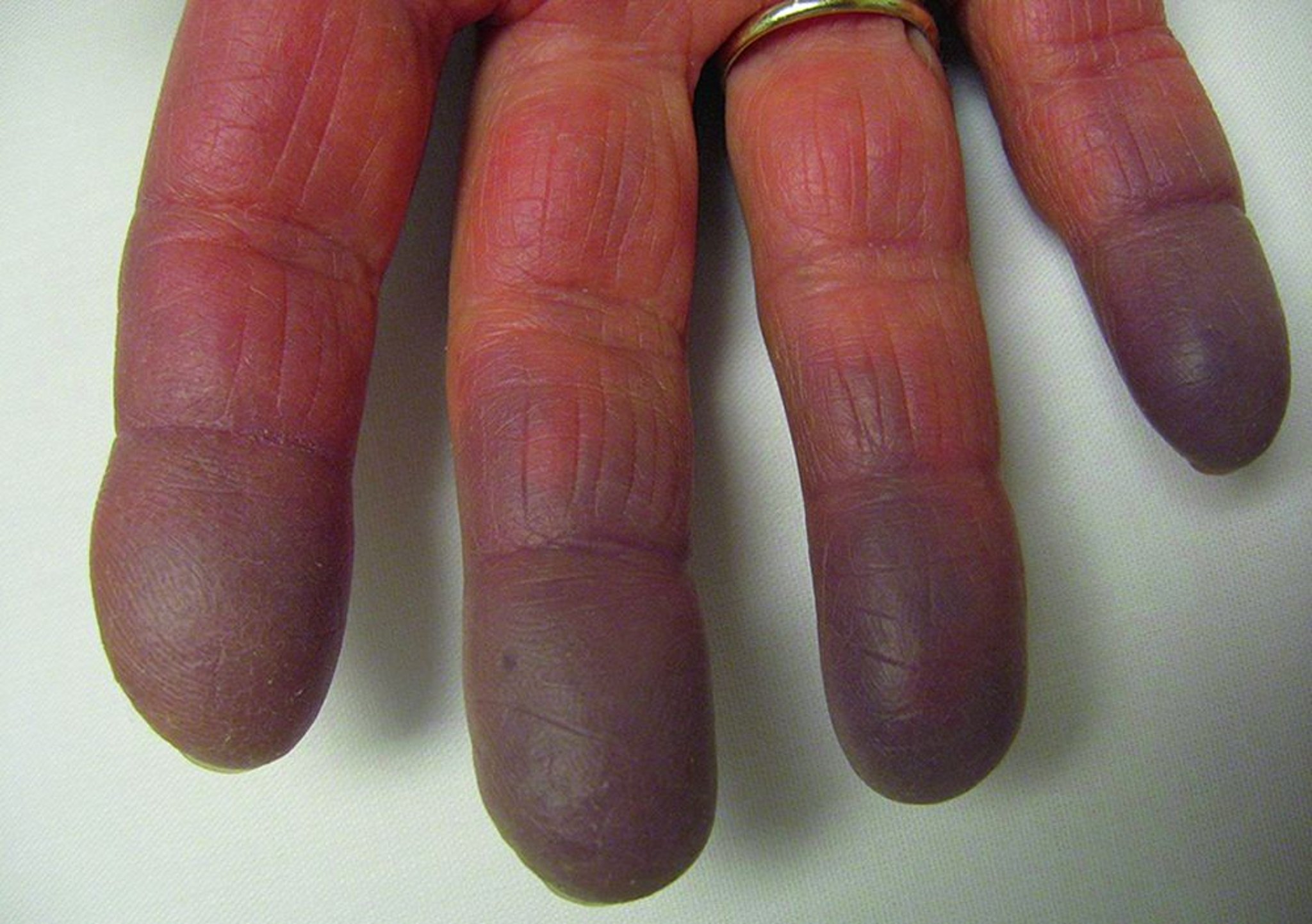
© Springer Science+Business Media
Hầu như tất cả các bệnh nhân đều bị đau nhiều khớp, và 75% bị viêm khớp. Viêm khớp thường không gây biến dạng khớp, nhưng tổn thương bào mòn khớp có thể thay đổi và có thể có biến dạng khớp giống như viêm khớp dạng thấp (ví dụ, biến dạng ngón tay cổ ngỗng hoặc ngón tay của người thợ thùa khuyết). Thường có yếu cơ gốc chi có hoặc không có co cứng cơ, đặc biệt ở những bệnh nhân có tăng men cơ (ví dụ như creatine kinase).
Tổn thương thận (thường gặp nhất là viêm cầu thận màng) gặp ở 25% số bệnh nhân và có mức độ nhẹ; tổn thương mức độ nặng hoặc có nguy cơ tử vong, là một thể không điển hình của bệnh mô liên kết hỗn hợp. Tổn thương phổi chiếm tới 75% bệnh nhân bị bệnh mô liên kết hỗn hợp. Bệnh phổi kẽ là tổn thương phổi hay gặp nhất; tăng áp lực động mạch phổi là một trong những nguyên nhân chính gây tử vong. Có thể có suy tim. Có thể có hội chứng Sjögren.
Chẩn đoán MCTD
Xét nghiệm kháng thể kháng nhân (ANA), kháng thể kháng kháng nguyên hòa tan nhân (kháng thể kháng U1 ribonucleoprotein, hoặc RNP), và Smith (Sm) và kháng thể kháng DNA.
Đánh giá tổn thương ở các cơ quan bằng triệu chứng lâm sàng.
Nên nghĩ tới bệnh mô liên kết hỗn hợp khi bệnh nhân có các triệu chứng lâm sàng chồng chéo của các bệnh Lupus ban đỏ hệ thống (SLE), xơ cứng bì hệ thống, hoặc viêm đa cơ.
Các xét nghiệm đối với ANA và kháng thể kháng kháng nguyên U1 RNP được chỉ định đầu tiên. Hầu như tất cả các bệnh nhân đều có độ chuẩn ANA huỳnh quang cao tạo ra hình ảnh đốm trên khay xét nghiệm. Có các kháng thể đối với U1 RNP, thường ở hiệu giá rất cao. Các kháng thể kháng Sm của kháng nguyên hòa tan nhân (kháng thể kháng Sm) và kháng thể kháng chuỗi xoắn kép DNA (âm tính trong bệnh mô liên kết hỗn hợp) được chỉ định để loại trừ các bệnh lý khác. Sự hiện diện của các kháng thể chống RNP là không đủ để chẩn đoán MCTD; các triệu chứng lâm sàng điển hình cũng được yêu cầu.
Yếu tố dạng thấp thường dương tính và có thể có chỉ số cao. Tốc độ máu lắng thường tăng cao.
Tăng áp lực động mạch phổi nên được phát hiện càng sớm càng tốt bằng đo chức năng phổi và siêu âm tim. Chỉ định thêm các thăm dò phụ thuộc vào triệu chứng và dấu hiệu của cơ quan tổn thương; biểu hiện của viêm cơ, tổn thương thận và phổi được phát hiện bằng các xét nghiệm và thăm dò đặc hiệu của các cơ quan này.
Creatine kinase, MRI, điện cơ hoặc sinh thiết cơ có thể giúp xác nhận sự hiện diện của viêm cơ như một phần của MCTD.
Tiên lượng về MCTD
Tỷ lệ sống sau 10 năm là khoảng 80%, nhưng tiên lượng phụ thuộc phần lớn vào những triệu chứng nặng. Bệnh nhân có các biểu hiện của xơ cứng bì toàn thể và viêm đa cơ có tiên lượng xấu hơn. Bệnh nhân có nguy cơ cao bị xơ vữa động mạch. Nguyên nhân gây tử vong bao gồm tăng áp lực động mạch phổi, suy thận, nhồi máu cơ tim, thủng đại tràng, nhiễm trùng lan tỏa và xuất huyết não. Một số bệnh nhân đạt được lui bệnh ổn định trong nhiều năm mà không cần điều trị.
Điều trị MCTD
Thuốc chống viêm không steroid (NSAID) hoặc thuốc chống sốt rét (ví dụ, hydroxychloroquine, chloroquine)
Corticosteroid và các thuốc ức chế miễn dịch khác (ví dụ, methotrexate, azathioprine, mycophenolate mofetil) đối với bệnh mức độ hoạt động vừa đến nặng
Thuốc chẹn kênh canxi (ví dụ, nifedipine) và chẹn phosphodiesterase (ví dụ, tadalafil) để điều trị hội chứng Raynaud
Điều trị toàn thể và liệu pháp điều trị ban đầu được điều chỉnh cho các biểu hiện lâm sàng đặc hiệu và có biểu hiện giống với lupus ban đỏ hệ thống hoặc các kiểu hình lâm sàng chiếm ưu thế. Hầu hết các bệnh nhân bị bệnh ở mức độ trung bình hoặc nặng đều đáp ứng với corticosteroid, đặc biệt nếu được điều trị sớm, và các thuốc ức chế miễn dịch khác (ví dụ, methotrexate, azathioprine, mycophenolate mofetil). Bệnh nhẹ thường được kiểm soát bởi NSAID, thuốc chống sốt rét (ví dụ, hydroxychloroquine, chloroquine) hoặc thỉnh thoảng là thuốc corticosteroid liều thấp. Tổn thương nặng ở các cơ quan quan trọng thường cần được điều trị bằng corticosteroid liều cao (ví dụ, prednisone 1 mg/kg/ đường uống 1 lần/ngày) và kết hợp các thuốc ức chế miễn dịch khác. Nếu bệnh nhân có biểu hiện các triệu chứng của viêm cơ hoặc xơ cứng bì hệ thống, cần phải điều trị các thể đó giống như các bệnh trên.
Bệnh nhân có hội chứng Raynaud nên được điều trị dựa trên các triệu chứng của họ và huyết áp của bệnh nhân dung nạp được các thuốc chẹn kênh canxi (ví dụ, nifedipine) và thuốc ức chế phosphodiesterase (ví dụ, tadalafil).
Tất cả bệnh nhân cần được sàng lọc chặt chẽ với xơ vữa động mạch. Bệnh nhân điều trị corticosteroid kéo dài nên được dự phòng loãng xương. Nếu sử dụng liệu pháp ức chế miễn dịch kết hợp, bệnh nhân nên được điều trị dự phòng nhiễm trùng cơ hội, chẳng hạn như Pneumocystis jirovecii (xem phòng ngừa viêm phổi do Pneumocystis jirovecii) và vắc-xin chống nhiễm trùng thông thường (ví dụ: viêm phổi liên cầu, cúm, COVID-19).
Một số chuyên gia khuyến khích sàng lọc để phát hiện tăng áp lực động mạch phổi định kỳ bằng đo chức năng hô hấp và/hoặc siêu âm tim 1 đến 2 năm một lần tùy theo các triệu chứng.
Những điểm chính
Bệnh mô liên kết hỗn hợp thường có biểu hiện giống với lupus ban đỏ hệ thống, xơ cứng bì toàn thể và/hoặc viêm đa cơ.
Điển hình, kháng thể ANA và kháng thể kháng U1 RNP dương tính; kháng thể kháng am và kháng thể kháng DNA âm tính, nhưng sự hiện diện của các kháng thể kháng RNP không đủ để chẩn đoán.
Dự đoán tăng áp lực động mạch phổi.
Điều trị bệnh thể nhẹ với NSAID hoặc thuốc chống sốt rét, và với thể nặng hơn điêu trị kết hợp corticosteroid với các thuốc ức chế miễn dịch.