



Амилоидозом называют группу различных состояний, характеризующихся отложением во внеклеточном пространстве нерастворимых фибрилл, состоящих из неагрегированных белков. Эти белки могут накапливаться локально, что проявляется относительно немногими симптомами, или во многих органах, вызывая тяжелую полиорганную недостаточность. Амилоидоз можеть развиваться как первичный (de novo) или вторичный, при различных инфекционных, воспалительных или злокачественных заболеваниях. Его диагностируют по результатам биопсии пораженной ткани; амилоидогенный белок типируют с помощью ряда иммуногистологических и биохимических методов. Лечение зависит от типа амилоидоза.
Амилоидные фибриллы состоят из нормально растворимых белков c неправильной пространственной укладкой, которые агрегируют с образованием олигомеров и затем нерастворимых фибрилл. К такому неправильному сворачиванию и агрегации чувствительны много нормальных (дикого типа) и мутантных белков (амилоидогенные белки), и в результате формируется спектр весьма разнообразных причин и типов амилоидоза.
Амилоидные отложения состоят из небольших (около 10 нм в диаметре) нерастворимых фибрилл, которые образуют конгофильные бета-складчатые листы, которые можно идентифицировать с помощью рентгеновской дифракции. Помимо фибриллярных белков, отложения амилоида содержат сывороточный Р-компонент и гликозаминогликаны.
Амилоидные отложения окрашиваются гематоксилином и эозином в розовый цвет, содержат углеводные компоненты, которые окрашиваются периодическим кислотнным (Шиффовым) красителем или альциановым синим, но наиболее характерно светло-зеленое двулучепреломление при микроскопии в поляризованном свете после окрашивания Конго красным. При аутопсии пораженные органы могут выглядеть восковидными.
Для развития амилоидоза, помимо выработки амилоидогенных белков, вероятно, важно также нарушение функционирования нормальных механизмов выведения таких неправильно сформированных белков. Сами по себе отложения амилоида метаболически инертны, но механически влияют на органы, препятствуя осуществлению их функций. Тем не менее, некоторые префибриллярные олигомеры амилоидогенных белков оказывают прямое цитотоксическое действие, что становится важным компонентом патогенеза заболевания.
Этиология амилоидоза
При системном амилоидозе циркулирующие амилоидогенные белки образуют отложения в различных органах. Основные системные типы включают
AL (первичный амилоидоз): обусловлен приобретенной избыточной экспрессией легких цепей клонального иммуноглобулина
AF (семейный амилоидоз): обусловлен наследованием мутантного гена, кодирующего белок с повышенным риском нарушения сворачивания, наиболее часто это транстиретин (TTR)
ATTRwt (генетический транститериновый амилоидоз дикого типа; ранее называемый старческим системным амилоидозом или ССА): обусловлен нарушением сворачивания и агрегации атипичного TTR
АА (вторичный амилоидоз): обусловлен агрегацией реагента острой фазы - сывороточного амилоида А
Амилоидоз, вызванный агрегацией бета-2-микроглобулина, может развиваться при хроническом гемодиализе, но благодаря использованию современных диализных мембран с большой пропускной способностью заболеваемость им снизилась. Существует редкая наследственная форма бета-2-микроглобулинового амилоидоза, обусловленная мутацией соответствующего гена.
Локализованные формы амилоидоза обусловлены, по-видимому, не осаждением циркулирующих белков, а местным синтезом и отложением амилоидогенного белка (чаще всего легких цепей иммуноглобулинов) в пределах пораженного органа. Часто пораженные участки включают центральную нервную систему (например, при болезни Альцгеймера), кожу, верхние или нижние дыхательные пути, паренхиму легких, мочевой пузырь, глаза и молочные железы.
AL амилоидоз (первичный амилоидоз)
AL амилоидоз обусловлен избыточной выработкой амилоидогенной легкой цепи иммуноглобулина при моноклональных плазмоклеточных или других В-лимфоцитарных лимфопролиферативных заболеваниях. Легкие цепи также могут образовывать в тканях нефибриллярные отложения (болезнь отложения легких цепей). Редко амилоидные фибриллы образуются из тяжелых цепей иммуноглобулина (так называемый амилоидоз AH).
Обычными местами таких отложений амилоида являются кожа, нервы, сердце, желудочно-кишечный тракт (включая язык), почки, печень, селезенка и кровеносные сосуды. Обычно в костном мозге наблюдается плазмацитоз низкой степени, аналогичный наблюдаемому при миеломной болезни, но в большинстве случаев истинная миеломная болезнь (с лизисом костей, гиперкальциемией, цилиндрурией и анемией) отсутствует. Однако в 10-20% случаев миеломной болезни развивается AL амилоидоз.
Амилоидоз AF (семейный амилоидоз)
AF вызван наследованием гена, кодирующего мутантный белок сыворотки с повышенной склонностью к агрегации; обычно этот белок активно вырабатывается печенью.
Сывороточные белки, которые могут вызвать AF, включают транстиретин (TTR), аполипопротеин А-I и А-II, лизоцим, фибриноген, гельзолин и цистатин С. Форма, предположительно семейная, вызванная сывороточным белком фактором хемотаксиса лейкоцитов 2 (LECT2); однако конкретная наследственная мутация гена для этого последнего типа окончательно не выявлена.
Наиболее распространенный тип AF- амилоидоз, вызванный TTR (ATTR). С амилоидозом связаны более 130 мутаций гена TTR. Наиболее распространенная мутация, V30M, часто встречается в Португалии, Швеции, Бразилии и Японии, а мутация V122I имеется приблизительно у 4% темнокожих Америки и стран Карибского бассейна. Пенетрантность и возраст развития заболевания весьма разнообразны, но стабильны в отдельных семьях и этнических группах (1).
ATTR вызывает периферическую сенсорную нейропатию, вегетативную нейропатию, хроническую болезнь почек, а также кардиомиопатию. Синдром запястного канала обычно предшествует другим неврологическим проявлениям болезни. За счет продукции эпителием сетчатки мутантного TTР могут формироваться гиалиновые отложения, или же могут накапливаться лептоменингеальные отложения, поскольку клетки хориоидного сплетения продуцируют мутантный TTР. Когда кардиомиопатия является преобладающим проявлением отложения ТТР в сердце, она называется транстиретин-амилоидной кардиомиопатией (АТТР-КМ).
ATTR-амилоидоз дикого типа (старческий системный амилоидоз)
ATTRwt вызван агрегацией и осаждениемTTR дикого типа, главным образом, в сердце.
Все чаще обнаруживают, что ATTR дикого типа вызывает инфильтративную кардиомиопатию у пожилых мужчин. Приблизительно 16% пациентов с аортальным стенозом, перенесших транскатетерную замену аортального клапана (2), и 13% пациентов, госпитализированных по поводу сердечной недостаточности, с сохраненной фракцией выброса (СНСФВ) также имеют транстиретиновую амилоидную кардиомиопатию, в данном случае обозначенную как wATTR-CM для обозначения отложения TTR дикого типа в сердце (3). Проявления в мягких тканеях ATTRwt амилоида, в том числе синдром запястного канала, разрыв межпальцевого сухожилия, разрыв ротаторной манжеты и спинальный стеноз, могут на годы предшествовать клиническому проявлению инфильтративной кардиомиопатии.
Генетические и эпигенетические факторы, вызывающие ATTRwt, неизвестны. Поскольку и ATTRwt, и AL амилоидоз могут вызывать развитие кардиомиопатии, и поскольку у больных в этой возрастной группе могут наблюдаться амилоидогенетические моноклональные гаммапатии, важно точно типировать амилоид, чтобы необоснованно не назначить при ATTRwt химиотерапию (которая используется для лечения AL).
АА амилоидоз (вторичній амилоидоз)
Эта форма амилоидоза развивается при различных инфекциях, воспалительных заболеваниях или злокачественных опухолях и обусловлена аггрегацией изоформ реактанта острой фазы воспаления – сывороточного амилоида А.
Распространенные причинные инфекции включают
Предрасполагающие для воспалительных процессов факторы включают
Наследуемые синдромы перемежающейся лихорадки, такие как семейная средиземноморская лихорадка
Болезнь Кастлмана
Воспалительные цитокины (например, интерлейкин [ИЛ]-1, фактор некроза опухоли [ФНО], ИЛ-6), вырабатываемые при этих заболеваниях или эктопически опухолевыми клетками, стимулируют образование в печени сывороточного амилоида А (SAA).
При АА амилоидозе поражаются в основном почки, селезенка, печень, надпочечники и лимфоузлы. Сердце или периферические и автономные нервы вовлекаются в патологический процесс на поздних этапах заболевания.
Локализованный амилоидоз
Локализованный амилоидоз за пределами головного мозга наиболее часто вызван отложением клональных легких цепей иммуноглобулина, внутри мозга преобладает амилоидный белок бета.
Локализованные отложения амилоида обычно отмечаются в дыхательных путях и легочной ткани, мочевом пузыре и мочеточниках, в коже, молочных железах, глазах. Редко амилоидоз вызывают другие белки, синтезируемые локально, такие как изоформы кератина, которые могут откладываться локально в коже. Клональные легкие цепи иммуноглобулинов, вырабатываемые ассоциированной со слизистой лимфоидной тканью в желудочно-кишечном тракте, дыхательных путях, мочевом пузыре, могут вызывать в этих органах локализованный AL.
Отложения бета-амилоидного белка в головном мозге способствуют развитию болезни Альцгеймера или цереброваскулярной амилоидной ангиопатии. У других белков, вырабатываемых в центральной нервной системе, также могут нарушаться образование складчатой структуры, происходить агрегация и повреждение нейронов, что приводит к развитию нейродегенеративных заболеваний (например, болезни Паркинсона, болезни Хантингтона).
Справочные материалы по этиологии
1. Buxbaum JN, Ruberg FL: Transthyretin V122I (pV142I)* cardiac amyloidosis: an age-dependent autosomal dominant cardiomyopathy too common to be overlooked as a cause of significant heart disease in elderly African Americans. Genet Med 19(7):733-742, 2017. doi:10.1038/gim.2016.200
2. Fabbri G, Serenelli M, Cantone A, et al: Transthyretin amyloidosis in aortic stenosis: clinical and therapeutic implications. Eur Heart J Suppl 23(Suppl E):E128-E132, 2021. doi:10.1093/eurheartj/suab107
3. Magdi M, Mostafa MR, Abusnina W, et al: A systematic review and meta-analysis of the prevalence of transthyretin amyloidosis in heart failure with preserved ejection fraction. Am J Cardiovasc Dis 12(3):102-111, 2022. PMID: 35873185
Симптомы и признаки амилоидоза
Симптомы и признаки системного амилоидоза неспецифичные, что часто приводит к задержкам в диагностике. Особенно высокое подозрение на амилоидоз возникает при прогрессирующих полисистемных заболеваниях.
Отложения амилоида в почках обычно возникают в мембраны клубочков, что приводит к развитию протеинурии, но примерно в 15% случаев поражаются канальцы, что проявляется азотемией с минимальной протеинурией. Эти процессы могут прогрессировать до развития нефротического синдрома с выраженной гипоальбуминемией, отеков, и анасарки или терминальной стадии почечной недостаточности.
При поражении печени наблюдается ее безболезненное, и иногда значительное, увеличение. Печеночные тесты, как правило, свидетельствуют о внутрипеченочном холестазе с высокими уровнями щелочной фосфатазы, а позже - билирубина, хотя желтуха развивается редко. В некоторых случаях развивается портальная гипертензия с варикозным расширением вен пищевода и асцитом.
Дыхательные пути и поражение гортани приводит к одышке, охриплости, визингу, кровохарканью или обструкции дыхательных путей.
Инфильтрация миокарда вызывает развитие рестриктивной кардиомиопатии, и в конечном итоге приводит к нарушению диастолической функции и развитию сердечной недостаточности; возможны внутрисердечные блокады или аритмии. Часто развивается гипотония.
AL-амилоидоз и АТТР часто проявляются периферической нейропатией с парестезиями в пальцах рук и ног. Вегетативная нейропатия может быть причиной ортостатической гипотензии, эректильной дисфункции, нарушения потоотделения, задержки мочи и нарушений моторики желудочно-кишечного тракта.
Цереброваскулярная амилоидная ангиопатия чаще приводит к развитию спонтанного мозгового кровоизлияния, но у некоторых пациентов неврологические симптомы могут быть короткими и транзиторными.
Отложения амилоида в желудочно-кишечном тракте могут нарушать моторику пищевода, тонкого и толстого кишечника. Возможны атония желудка, нарушения процессов всасывания, кровотечения или кишечная псевдонепроходимость. При AL-амилоидозе обычно наблюдается макроглоссия.
Инфильтрация амилоида в мягких тканях обычно предшествует клинической экспрессии ATTRwt-амилоидной кардиомиопатии. Манифестация амилоидоза мягких тканей включает в себя карпальный туннельный синдром, синдром щёлкающего пальца, разрыв сухожилия бицепса и стеноз спинномозгового канала.

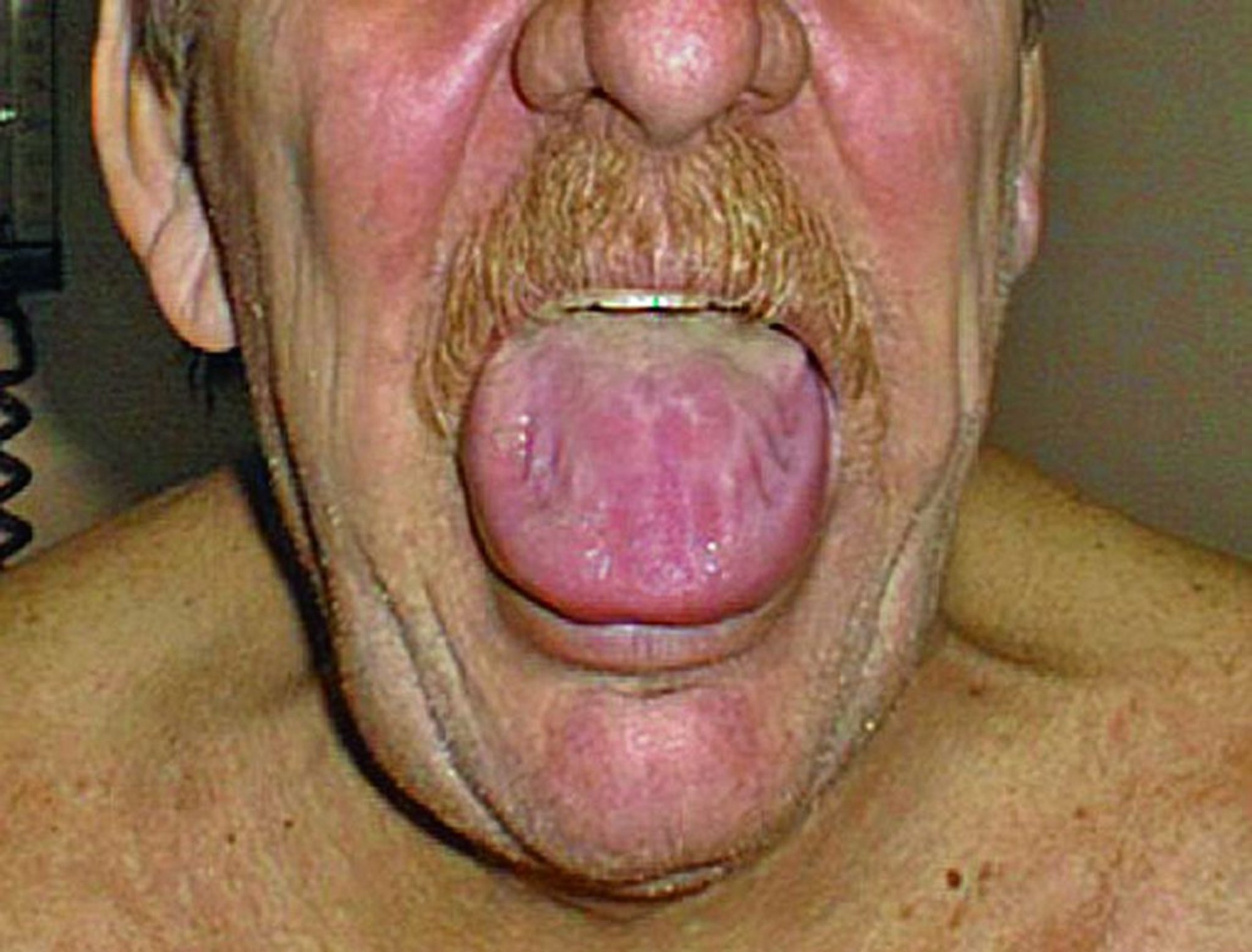
© Springer Science+Business Media
Амилоидоз щитовидной железы может стать причиной уплотненного, симметричного, безболезненного зоба, похожего на тиреоидит Хашимото. Также могут возникать другие эндокринопатии.
Поражение легких (главным образом при AL-амилоидозе) может характеризоваться очаговыми легочными узелками и кистами, патологическими изменениями в трахеобронхиальном дереве, плевральным выпотом или диффузными альвеолярно-септальными (интерстициальными) отложениями.
Амилоид стекловидной непрозрачности и двусторонние фестончатые колебания зрачков развиваются при нескольких наследственных амилоидозах.
Другими проявлениями являются кровоподтеки, часто вокруг глаз ("глаза енота"), обусловленные отложением амилоида в кровеносных сосудах. Амилоидные отложения вызывают ослабление кровеносных сосудов, что может привести к их разрыву после незначительной травмы, такой как чихание или кашель.
Диагностика амилоидоза
Биопсия
Типирование амилоида
Тестирование для выявления органных поражений
Биопсия
Амилоидоз диагностируют при выявлении в пораженных органах фибриллярных отложений. Результат исследования аспирационного биоптата подкожной жировой клетчатки обнаруживает амилоидные отложения в 80% случаев у пациентов с AL типом амилоидоза, но менее чем в 25% случаев при ATTRwt типе (1). Если результат биопсии подкожной жировой клетчатки отрицательный, следует провести биопсию клинически пораженного органа. Диагностическая чувствительность биопсии почек и сердца составляет почти 100%, если эти органы поражены клинически. Срезы тканей окрашивают Конго красным и исследуют под поляризационным микроскопом для выявления характерного двойного лучепреломления. Неветвящиеся 10-нм фибриллы также можно выявить с помощью электронной микроскопии в биоптатах ткани сердца или почек.
Ядерное сканирование с использованием трейсеров, тропных к костной ткани, может подтвердить диагноз транстиретиновой семейной амилоидной кардиомиопатии (ATTR) без биопсии сердца, при условии, что AL-амилоидоз исключен.
Типирование амилоида
После подтверждения диагноза амилоидоза с помощью биопсии определяют его тип, для чего предложен ряд методов. Для некоторых типов амилоидоза диагностически значима может быть иммуногистохимия или иммунофлюоресценция, но возможны и ложно-положительные результаты типирования. Другие полезные методы включают секвенирование генов при AF и биохимическую идентификацию с помощью масс-спектрометрии для точной идентификации вариантов белка в амилоидных отложениях (наиболее чувствительный и специфический метод).
Если заподозрен AL, необходимо исключать основное заболевание с участием плазматических клеток, для чего проводят количественное измерение содержания легких цепей иммуноглобулинов в бессывороточном препарате, качественное определение моноклональных легких цепей в сыворотке или моче с использованием иммунофиксации и электрофореза (электрофорез белков сыворотки и электрофорез белков мочи при AL нечувствительны) и биопсию костного мозга с проточной цитометрией или иммуногистохимическим исследованием для выявления клональности плазматических клеток.
При выявлении > 10% клоновых плазматических клеток необходимо дообследование, чтобы оценить соответствие критериям множественной миеломы, в том числе с проведением скрининга на литические поражения костей, анемию, почечную недостаточность и гиперкальциемию.
Поражение органов
Пациентов обследуют для выявления поражения внутренних органов, начиная с неинвазивного исследования:
Почки: анализ мочи; измерение уровней АМК, креатинина и альбумина; расчетной скорости клубочковой фильтрации (рСКФ); и 24-часовый сбор мочи для электрофореза белков (СМЭБ)
Печень: функциональные тесты печени
Легкие: рентгенография грудной клетки, КТ грудной клетки и легочные функциональные пробы
Сердце: ЭКГ и измерение уровня биомаркеров, таких как мозгового (В-типа) натрийуретического пептида (BNP) или N-терминального про-BNP (NT-proBNP) и тропонина
Поражение сердца можно заподозрить при низком вольтаже ЭКГ (вызванном утолщением стенки желудочка) и/или аритмиях. Если из-за симптомов подозревается вовлечение сердца, в дополнение к данным ЭКГ и сердечным биомаркерам проводится эхокардиография для измерения диастолического расслабления и общей продольной деформации (показатель систолической функции левого желудочка), а также для скрининга бивентрикулярной гипертрофии. В неоднозначных случаях может быть проведена МРТ сердца для выявления стойкого субэндокардиального усиления накопления гадолиния, что является характерным признаком заболевания. Сцинтиграфия миокарда с пирофосфатом технеция улучшила обнаружение ATTR-амилоидоза сердца и может помочь избежать необходимости биопсии сердца, при условии, что анализы крови исключают AL-амилоидоз (2, 3).
Справочные материалы по диагностике
1. Aimo A, Emdin M, Musetti V, et al: Abdominal Fat Biopsy for the Diagnosis of Cardiac Amyloidosis. JACC Case Rep 2(8):1182-1185, 2020. doi:10.1016/j.jaccas.2020.05.062
2. Gillmore JD, Maurer MS, Falk RH, et al: Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation 133(24):2404–2412, 2016.
3. Maurer MS, Bokhari S, Damy T, et al: Expert consensus recommendations for the suspicion and diagnosis of transthyretin cardiac amyloidosis. Circ Heart Fail 12(9):e006075, 2019.
Лечение амилоидоза
Поддерживающая терапия
Типоспецифическое лечение
Существуют специфические методы лечения большинства форм амилоидоза, хотя некоторые из них находятся на стадии исследований. При всех формах системного амилоидоза облегчить симптомы и улучшить качество жизни можно с помощью поддерживающих мероприятий.
Поддерживающая терапия
Поддерживающую терапию подбирают в зависимости от пораженной системы органов:
Почки: при нефротическом синдроме и отеках следует ограничить потребление соли и жидкости, и назначить петлевые диуретики; из-за продолжающейся потери белка потребление белка не ограничивают. Трансплантацию почки обсуждают, если удается контролировать течение основного заболевания; она может обеспечить длительное выживание, сравнимое с достигаемым при других заболеваниях почек.
Сердце: при кардиомиопатии следует ограничить потребление соли и жидкости, и назначить петлевые диуретики. Другие препараты при сердечной недостаточности, в том числе дигоксин, ингибиторы ангиотензинпревращающего фермента (АПФ), блокаторы кальциевых каналов и бета-блокаторы плохо переносятся и противопоказаны. В отдельных тщательно подобранных случаях AL или ATTR амилоидоза с поражением сердца с успехом проводилась пересадка сердца. Для предотвращения рецидивов в трансплантированном сердце пациентам с амилоидозом AL должна быть назначена агрессивная химиотерапия, направленная на лечение нарушений со стороны клональных плазматических клеток, а для пациентов с симптоматической амилоидной полинейропатией (САП) или с кардиомиопатией следует рассмотреть возможность анти-TTR терапии.
Желудочно-кишечный тракт: при диарее может быть полезен лоперамид. При раннем насыщении и замедленном опорожнении желудка может быть эффективен метоклопрамид.
Нервная сиситема: при периферической нейропатии боль могут облегчить габапентин, дулоксетин или прегабалин.
Ортостатическая гипотензия часто уменьшается при терапии высокими дозами мидодрина; у пожилых мужчин этот препарат может вызывать задержку мочи, однако лекарственные осложнения при гипертонии в положении лежа на спине у данной группе пациентов редки. Также можно носить противоотечные чулки, а в отсутствие периферических отеков, анасарки или сердечной недостаточности можно назначить флудрокортизон. Мидодрин, флудрокортизон или дроксидопа могут быть добавлены у пациентов с рефрактерной ортостатической гипотензией.
AL амилоидоз
Для AL амилоидоза
Для сохранения функции органов и увеличения продолжительности жизни важно быстро начать терапию для подавления активности плазматических клеток.
Большинство препаратов, применяемых для лечения множественной миеломы, использовались для лечения AL-амизоидоза; выбор препарата, дозы и схемы введения часто приходится изменять при нарушениях функции органов.
Первой схемой, для которой удалось выявить какую-либо эффективность, стала химиотерапия с использованием алкилирующего препарата (например, мелфалана, циклофосфамида) в сочетании с кортикостероидами. Высокие дозы мелфалана в/в в сочетании с трансплантацией аутологичных стволовых клеток могут в некоторых случаях оказаться высокоэффективны (1).
Также могут быть эффективны ингибиторы протеасомы (например, бортезомиб) и иммуномодуляторы (например, леналидомид). Исследование применения моноклональных антител даратумумаба в сочетании с циклофосфамидом, бортезомибом и дексаметазоном у пациентов с недавно диагностированным AL-амилоидозом (за исключением пациентов с сердечной недостаточностью классов III и IV по NYHA, уровнем N-терминального фрагмента про-В натрийуретического белка [NTproBNP] > 8500 пг/мл [> 1003 пмоль/л] и рСКФ < 20 мл/мин/м2) показало беспрецедентно высокий уровень гематологического ответа (2). Гематологический ответ основан на наличии моноклонального белка в сыворотке и моче, определяемом с помощью электрофореза с иммунофиксацией, и на уровнях легких цепей сыворотки в соотношении каппа/лямбда. Однако данные о долгосрочной выживаемости отсутствуют.
Все доступные методы лечения направлены на клональные В-клетки или плазматические клетки при AL-амилоидозе. Исследования антифибриллярных антител, таких как биртамимаб и CAEL-101, продолжаются (3).
Локализованный AL-амилоидоз можно лечить низкими дозами наружной дистанционной лучевой терапии, поскольку плазматические клетки весьма радиочувствительны.
ATTR-амилоидоз
При ATTR-амилоидозе назначаются:
Трансплантация печени
Тетрамер-стабилизирующие препараты
Генные глушители
Трансплантация печени, которая заменяет первичное место синтеза мутантного белка новым органом, производящим нормальный ТТР, может быть эффективной при некоторых мутациях TTR если она проводится в начале заболевания (ранняя нейропатия и отсутствие поражения сердца). Трансплантация на более поздних стадиях заболевания часто приводит к прогрессирующей амилоидной кардиомиопатии и нейропатии из-за неправильного скручивания и отложения белка TTR дикого типа на уже существующие отложения амилоида.
Было показано, что несколько препаратов позволяют стабилизировать уровень циркулирующих тетрамеров TTР в плазме, ингибируют неправильное скручивание TTР и образование фибрилл, а также эффективно замедляют неврологическое прогрессирование заболевания, сохраняя качество жизни. К стабилизаторам TTR относятся дифлунизал, широко доступный генерический противовоспалительный препарат, и тафамидис (4, 5).
Подавление гена TTR с использованием анти-смысловой РНК или РНК-интерференции для блокировки трансляции TTR мРНК эффективно снижает уровни TTR в сыворотке, улучшая результаты неврологического лечения у 50% пациентов и, по всей видимости, способно восстанавливать поврежденные нервные волокна (6, 7). Доступны препараты, подавляющие гены: патисиран, инотерсен и вутрисиран.
Исследование вутрисирана, генного глушителя второго поколения, продемонстрировало улучшение функциональных результатов у пациентов с семейной амилоидной полинейропатией (8). Предварительные данные другого исследования показывают, что генные глушители могут быть эффективны при лечении кардиомиопатии у пациентов с АТТР-амилоидозом (9).
ATTR-амилоидоз дикого типа
Для ATTR-амилоидоза дикого типа
Тетрамер-стабилизирующие препараты
Было показано, что стабилизация TTR с использованием тафамидиса у пациентов с ATTR или ATTRw-амилоидной кардиомиопатией снижает смертность от всех причин и число госпитализаций, связанных с сердечно-сосудистыми заболеваниями (5). Клинические испытания в настоящее время изучают влияние глушителей гена TTR на кардиомиопатию, которая возникает у пациентов с амилоидозом АТТР дикого типа, а также на кардиомиопатию, которая возникает у пациентов с амилоидозом АТТР, характеризующимся мутантным белком (10).
В отличие от наследственного ATTR-амилоидоза, трансплантация печени не эффективна у пациентов с ATTR-амилоидозом дикого типа, поскольку амилоидный белок по своей структуре является генетически нормальным транстиретином (TTР).
АА амилоидоз
При АА амилоидозе, вызванном семейной средиземноморской лихорадкой, эффективен пероральный колхицин.
Лечение других типов АА направлено на купирование основного заболевания: инфекции, воспалительного заболевания или рака.
Для прерывания передачи сигнала от цитокинов могут использоваться колхицин или анти-ИЛ-1-, анти-ИЛ-6- или анти-ФНО-препараты, при этом они уменьшают воспалительный процесс, приводящий к продукции сывороточного амилоида A (СAA) в печени.
Справочные материалы по лечению
1. Sanchorawala V, Sun F, Quillen K, et al: Long-term outcome of patients with AL amyloidosis treated with high-dose melphalan and stem cell transplantation: 20-year experience. Blood 126: 2345–2347, 2015. doi: 10.1182/blood-2015-08-662726
2. Kastritis E, Palladini G, Minnema MC, et al: Daratumumab-Based Treatment for Immunoglobulin Light-Chain Amyloidosis. N Engl J Med 385(1):46-58, 2021. doi:10.1056/NEJMoa2028631
3. Quarta CC, Fontana M, Damy T, et al: Changing paradigm in the treatment of amyloidosis: From disease-modifying drugs to anti-fibril therapy. Front Cardiovasc Med 9:1073503, 2022. doi:10.3389/fcvm.2022.1073503
4. Berk JL, Suhr OB, Obici L, et al: Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA 310: 2658–2667, 2013. doi: 10.1001/jama.2013.283815
5. Maurer MS, Schwartz JH, Gundapaneni B, et al: Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med 379:1007–1016, 2018.
6. Adams D, Gonzalez-Duarte A, O'Riordan WD, et al: Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med 379:11–21, 2018.
7. Benson MD, Waddington-Cruz M, Berk JL, et al: Inotersen treatment for patients with transthyretin amyloidosis. N Engl J Med 379:22–31, 2018.
8. Adams D, Tournev IL, Taylor MS, et al: Efficacy and safety of vutrisiran for patients with hereditary transthyretin-mediated amyloidosis with polyneuropathy: a randomized clinical trial. Amyloid 30(1):1-9, 2023. doi:10.1080/13506129.2022.2091985
9. Maurer MS, Fontanta MA, Berk JL, et al: Primary results from APOLLO-B, a phase 3 study of patisiran in patients with transthyretin-mediated amyloidosis with cardiomyopathy. Abstract presented at International Symposium of Amyloidosis, September 2022,Heidelberg Germany.
10. Writing Committee, Kittleson MM, Ruberg FL, et al: 2023 ACC Expert Consensus Decision Pathway on Comprehensive Multidisciplinary Care for the Patient With Cardiac Amyloidosis: A Report of the American College of Cardiology Solution Set Oversight Committee [published correction appears in J Am Coll Cardiol 81(11):1135, 2023]. J Am Coll Cardiol 81(11):1076-1126, 2023. doi:10.1016/j.jacc.2022.11.022
Прогноз при амилоидозе
Прогноз зависит от типа амилоидоза и пораженной системы органов, но при соответствующем патогенетическом лечении и поддерживающей терапии продолжительность жизни многих пациентов достаточно велика.
AL амилоидоз осложняется тяжелой кардиомиопатией, и его прогноз остается наименее благоприятным, медиана выживаемости составляет < 1 г. Нелеченый ATTR-амилоидоз, как правило, в течение 5-15 лет прогрессирует до терминальной стадии поражения сердца или нервной системы. Считалось, что амилоид дикого типа ATTR обычно прогрессирует медленней остальных типов системного амилоидоза, которые поражают сердце, но у пациентов с амилоидом дикого типа ATTR заболевание прогрессирует в течение медианы 4-х лет от момента взятия биопсии и постановки диагноза до развития клинически проявляющейся сердечной недостаточности и смерти.
Прогноз при АА амилоидозе во многом зависит от эффективности лечения основного инфекционного, воспалительного или опухолевого заболевания.
Основные положения
Амилоидоз представляет собой группу заболеваний, при которых нарушается образование складок молекулы определенных белков, которые агрегируют с образованием нерастворимых фибрилл, откладывающихся в органах, нарушая их функцию.
Нарушение образования складок выявлено у многих различных белков; некоторые из этих белков вырабатываются при генетическом дефекте или при определенных патологических состояниях, тогда как другие являются легкими цепями иммуноглобулинов, вырабатываемыми моноклональными плазматическими клетками или другими В-лимфоцитами при лимфопролиферативных нарушениях.
Тип амилоида и клиническое течение заболевания определяется типом амилоидогенного белка, хотя клинические проявления различных типов амилоидоза могут перекрываться.
Могут поражаться многие органы, особенно неблагоприятным прогнозом сопровождается поражение сердца; амилоидная кардиомиопатия, как правило, приводит к нарушению диастолической функции, развитию сердечной недостаточности и внутрисердечной блокады или аритмии.
Диагноз устанавливают по данным биопсии; тип амилоидоза определяют различными иммунологическими, генетическими и биохимическими методами. Масс-спектрометрия представляет собой наиболее чувствительный и специфичный метод для типирования амилоида.
Облегчить симптомы и улучшить качество жизни помогает соответствующее поддерживающая терапия; в отдельных случаях может быть эффективна трансплантация органов.
Лечат основной патологический процесс; для AL амилоидоза вследствие плазмоклеточных или лимфопролиферативных заболеваний может быть высокоэффективна химиотерапия; для вторичного АА амилоидоза - противоинфекционные и противовоспалительные препараты могут помочь.
При наследственном ATTR-амилоидозе низкомолекулярные стабилизирующие лекарственные средства и препараты, вызывающие выключение гена, подавляют или, возможно, обращают вспять неврологическое ухудшение; у пациентов с амилоидной кардиомиопатией (ATTR или ATTRwt) применение тафамидиса снижает общую летальность и количество случаев госпитализаций по причине сердечно-сосудистых расстройств.







