



Аутоиммунный миозит характеризуется воспалительными и дегенеративными изменениями мышц (полимиозит, некротизирующая иммуно-опосредованная миопатия) либо мышц и кожи (дерматомиозит). Проявления включают симметричную слабость, изредка чувствительность, замещение мышцы фиброзной тканью, иногда с развитием атрофии, в основном проксимальных мышц тазового и плечевого пояса. Диагноз ставится на основании клинических данных и патологических результатов мышечных тестов, которые могут включать тест на креатинкиназу, МРТ, электромиографию и биопсию мышц. При некоторых типах миозита имеются симптомы нарушений в работе легких и сердца. При лечении используют кортикостероиды в комбинации с иммунодепрессантами и/или внутривенным введением иммуноглобулина.
Аутоиммунный миозит в 2 раза чаще встречается у женщин, чем у мужчин. Заболеваемость у людей с темной кожей в 3-4 раза выше, чем у людей с белой кожей. Заболевание может встречаться в любом возрасте, но чаще выявляется в интервале от 40 до 60 лет, у детей – от 5 до 15 лет.
Этиология аутоиммунного миозита
Считают, что причиной аутоиммунного миозита может быть аутоиммунная реакция на мышечную ткань у генетически предрасположенных лиц. Возникает семейная склонность к заболеванию, а подтипы лейкоцитарного антигена человека (HLA) связаны с миозитом. Например, аллели 8,1-го наследственного гаплотипа (HLA-DRB1 * 03-DQA1 * 05-DQB1 * 02) увеличивают риск заболевания полимиозитом, дерматомиозитом и интерстициальными заболеваниями легких. Возможными пусковыми факторами являются вирусные миозиты и злокачественные ново-образования. Ассоциация злокачественных опухолей с дерматомиозитом (в меньшей степени с полимиозитом) позволяет предположить, что опухоли могут являться пусковым механизмом развития заболевания в результате возникновения аутоиммунных реакций на общие антигены опухоли и мышечной ткани.
Патофизиология аутоиммунного миозита
Патологические изменения включают повреждение клеток и их атрофию на фоне воспаления различной степени выраженности. Мышцы кистей, стоп и лица повреждаются в меньшей степени, чем другая скелетная мускулатура. Поражение мускулатуры глотки и верхних отделов пищевода, реже – сердечной мышцы может приводить к нарушению функций указанных органов. Также возможны воспалительные изменения суставов и легких, особенно при наличии антисинтетазных антител.
Дерматомиозит характеризуется отложением иммунных комплексов в стенках сосудов и рассматривается как опосредованная комплементом васкулопатия. Напротив, полимиозит характеризуется прямым Т-клеточно-опосредованным повреждением мышц, а некротизирующие иммуноопосредованные миопатии характеризуются инфильтратами с преобладанием макрофагов и миофагоцитозом.
Классификация аутоиммунного миозита
Аутоиммунный миозит можно разделить на 4 группы, в основном опираясь на данные гистопатологии и клинические проявления:
Полимиозит
Дерматомиозит
Некротизирующие иммуно-опосредованные миопатии
Миозит с включенными тельцами
Дерматомиозит можно отличить от полимиозита по характерным кожным симптомам дерматомиозита (см. Симптомы и признаки). Мышечная гистопатология также отличается. Дерматомиозит и полимиозит могут проявляться как чисто мышечные заболевания или как часть антисинтетазного синдрома, который может быть ассоциирован с артритом (обычно неэрозивным), лихорадкой, интерстициальной болезнью легких, гиперкератозом радиальной поверхности пальцев рук (руки механика) и синдромом Рейно.
Некротизирующие иммуно-опосредованные миопатии чаще всего включают в себя миозит, ассоциированный с антителами к сигнал-узнающей частице (SRP), и статин-индуцированный миозит, обычно имеют агрессивные проявления, с очень высокими уровнями креатинкиназы (КК) и не поражают экстрамышечные органы (1).
Миозит с тельцами включения вызывает слабость проксимальных мышц ног, но часто поражает дистальные мышцы (например, мышцы рук и ног), часто сопровождающиеся их атрофией. Он развивается в более старшем возрасте, имеет более медленное прогрессирование и, как правило, не отвечает на иммуносупрессивную терапию.
Аутоиммунный миозит может также накладываться на другие аутоиммунные расстройства, например, системную красную волчанку, системный склероз, смешанные заболевания соединительной ткани. У таких пациентов в дополнение к миозиту наблюдаются симптомы и признаки перекрывающихся заболеваний (проявляются как дерматомиозит или полимиозит).
Справочные материалы по классификации
1. Lundberg IE, Fujimoto M, Vencovsky J, et al. Idiopathic inflammatory myopathies. Nat Rev Dis Primers 7(1):86, 2021. doi:10.1038/s41572-021-00321-x
Симптомы и признаки аутоиммунного миозита
Начало аутоиммуного миозита может быть острым (особенно у детей) или постепенным (особенно у взрослых). Он может также сопровождаться полиартралгиями, синдромом Рейно, дисфагией, легочными проявлениями (например, кашель, одышка), общими симптомами (лихорадка, утомляемость, снижение массы тела). Тяжелая степень заболевания характеризуется дисфагией, дисфонией и/или слабостью диафрагмы.
Мышечная слабость может прогрессировать в течение нескольких недель или месяцев. Для клинического проявления мышечной слабости необходимо поражение минимум 50% мышечных волокон (таким образом, наличие мышечной слабости свидетельствует о наличии развернутой стадии миозита). Пациенты могут испытывать затруднения при подъеме рук выше уровня плеч, ходьбе вверх по лестнице, подъеме из положения сидя. Иногда развивается болезненность мышц и их атрофия. Вследствие выраженной слабости мышц плечевого и тазового пояса пациентам может потребоваться использование кресла-каталки или они могут стать прикованными к постели. При поражении сгибателей шеи становится невозможным оторвать голову от подушки. Поражение мышц глотки и верхних отделов пищевода приводит к нарушению глотания и аспирации пищи. Мышцы кистей, стоп и лица не поражаются, за исключением миозита с включенными тельцами, при котором характерной является вовлеченность дистальных отделов, особенно кистей. Контрактуры конечностей развиваются редко.
Суставные проявления включают полиартралгию или полиартрит, с припухлостью и другими признаками недеформирующего артрита. Они чаще наблюдаются при наличии антител к Jо-1 или других антисинтетазных антител.
Поражение внутренних органов, за исключением глотки и верхних отделов пищевода, при аутоиммунном миозите встречается реже, чем при других ревматических заболеваниях (например, при системной красной волчанке и системной склеродермии). Иногда, особенно при антисинтетазном синдроме, болезнь наиболее выраженно проявляется в виде интерстициального заболевания легких (с одышкой и кашлем). Могут возникать поражения сердца сердечные аритмии, особенно нарушения проводимости и дисфункция желудочка. Желудочно-кишечные симптомы, более характерные для детей, обусловлены ассоциацией с васкулитом и могут проявляться рвотой с кровью, меленой и ишемической перфорацией кишечника.
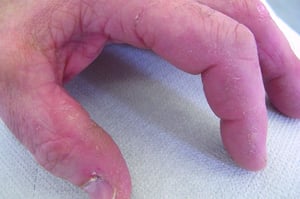
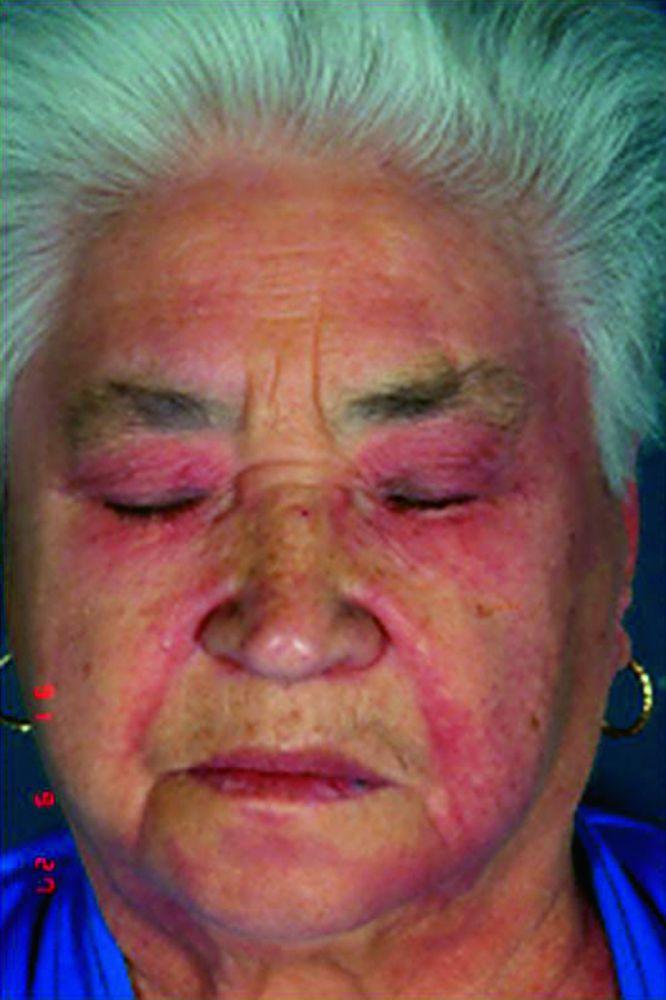
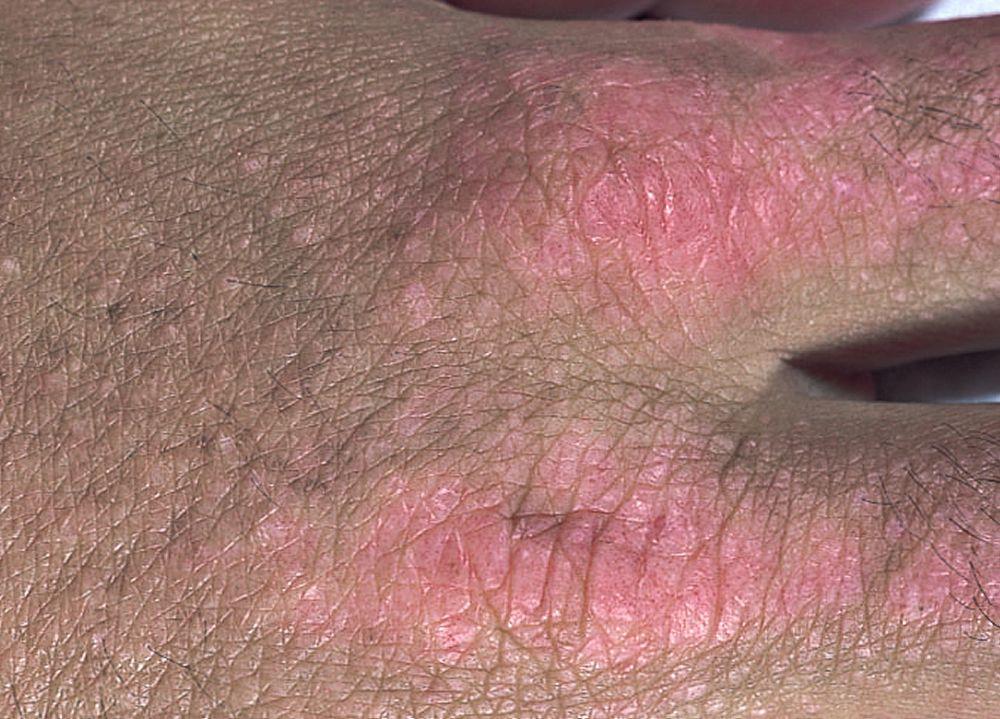
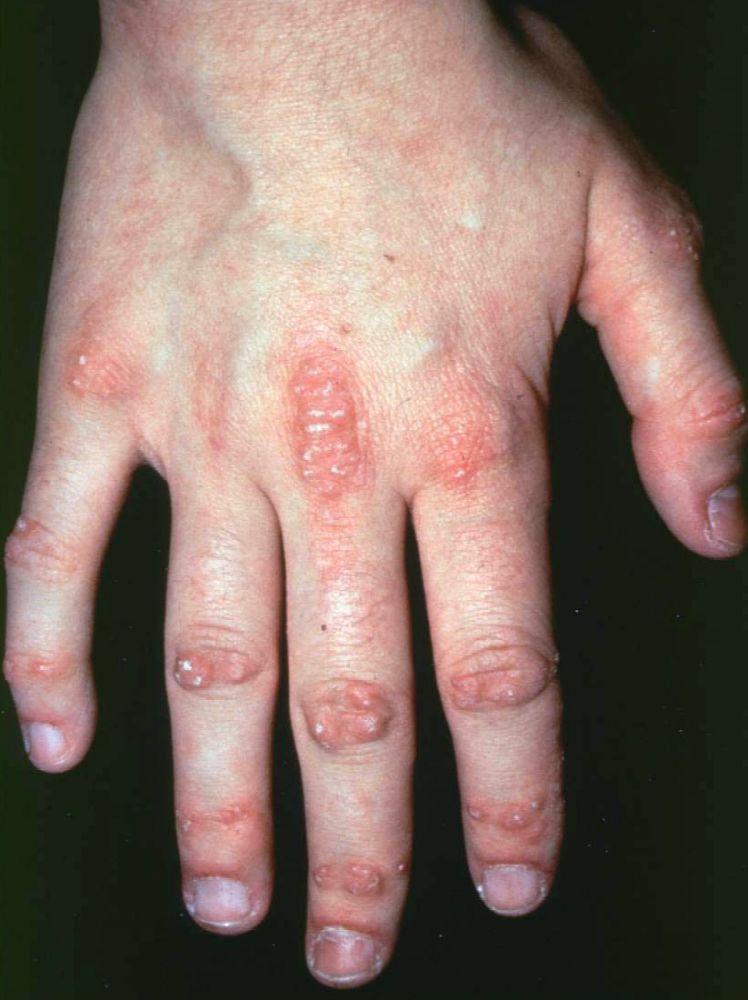
Кожные изменения, отмечающиеся при дерматомиозите, обычно имеют темный цвет и эритематозный характер. Выявляется фоточувствительность и изъязвление кожи. Относительно специфическим признаком дерматомиозита является периорбитальный отек пурпурного цвета (гелиотропная сыпь). При других локализациях кожные высыпания могут немного возвышаться над уровнем кожи и быть гладкими или покрытыми чешуйками; локализация высыпаний – лоб, шея в пределах V-образного выреза, плечи, грудь, спина, предплечья, нижние отделы ног, латеральные части бедер, локти и колени, медиальная часть лодыжек и тыльные поверхности проксимальных межфаланговых и пястно-фаланговых суставов (симптом Готтрона – относительно специфический признак). Возможна гиперемия или утолщение основания и периферии ногтей. На коже латеральной поверхности пальцев возможно развитие десквамативного дерматита, сопровождающегося появлением трещин. Возможно образование подкожных и мышечных кальцинатов, особенно у детей. Первичные кожные изменения часто разрешаются полностью, но могут приводить к развитию вторичных нарушений в виде темной пигментации, атрофии, стойкой неоваскуляризации, рубцов. Интенсивно зудящая сыпь на коже головы может напоминать псориаз.
Могут возникать характерные изменения кожи при отсутствии мышечного заболевания, и в таком случае болезнь называется амиопатическим дерматомиозитом.
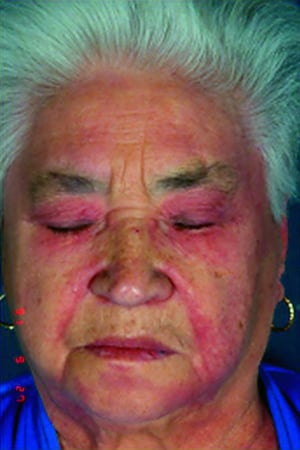
© Springer Science+Business Media
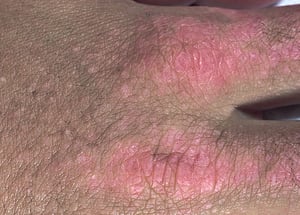
© Springer Science+Business Media
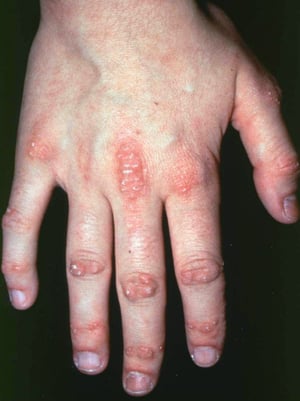
© Springer Science+Business Media
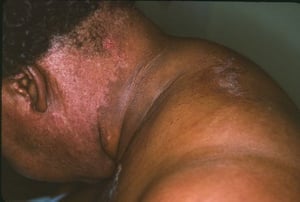
Image courtesy of Karen McKoy, MD.
© Springer Science+Business Media

© Springer Science+Business Media

© Springer Science+Business Media

© Springer Science+Business Media

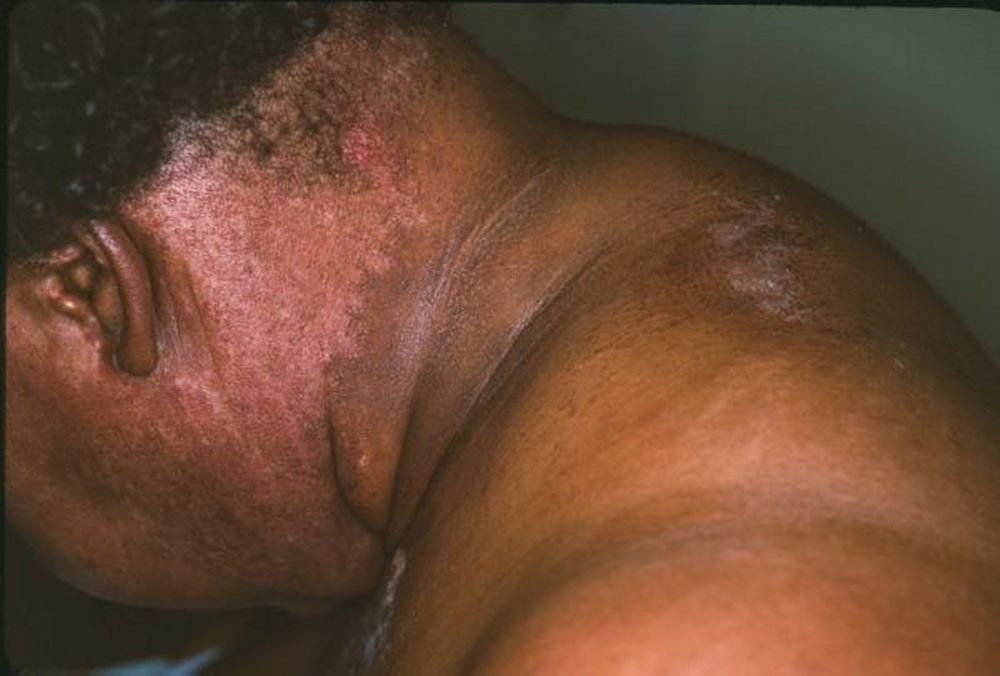
Image courtesy of Karen McKoy, MD.

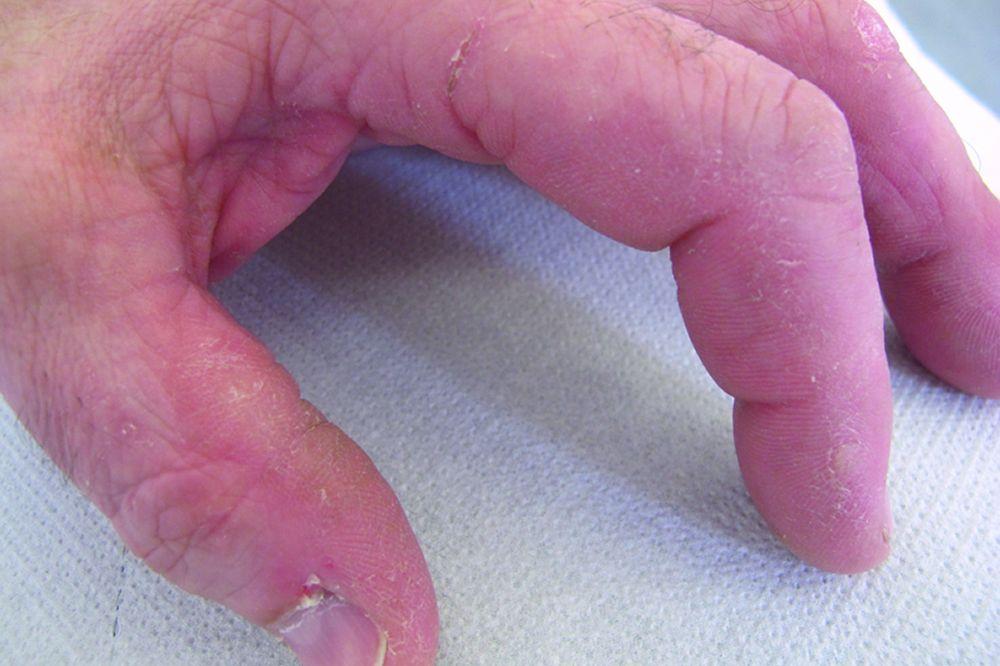
© Springer Science+Business Media
Диагностика аутоиммунного миозита
Клинические критерии
Биопсия мышцы (определяющая)
Аутоиммунный миозит следует заподозрить при наличии у пациентов слабости проксимальных мышц, с болезненностью или без нее. Дерматомиозит следует подозревать у пациентов с симптомами миозита и кожными поражениями, соответствующими дерматомиозиту. Для постановки диагноза аутоиммунного миозита необходимо наличие как можно большего количества из следующих 5 критериев:
слабость проксимальных мышц;
характерная сыпь;
Повышение уровня мышечных ферментов в сыворотке (если уровень креатинкиназы [КК] не повышен, необходимо исследовать уровни аминотрансфераз или альдолазы, которые намного менее специфичны, чем КК);
мышечные нарушения, выявляемые при электромиографии или МРТ;
изменения, выявляемые при биопсии мышцы (определяющий тест)
Изменения, выявляемые при биопсии, различаются, однако типичными являются хроническое воспаление с очагами дегенерации и некоторой регенерации мышечной ткани. Полимиозит и дерматомиозит часто можно распознать с помощью биопсии мышц. Перед началом лечения полимиозита необходимо поставить точный диагноз с помощью биопсии мышц для исключения других заболеваний мышц, таких, как заболевания в связи с отсутствием или недостатка ферментов, некротический миозит и поствирусный рабдомиолиз. Если кожные симптомы характерны для дерматомиозита, то биопсия мышц обычно не требуется. При дерматомиозите при биопсии не обнаруживается патогномоничных кожных признаков, но отсутствие прямой иммунофлюоресценции помогает отличить сыпь от сыпи у пациентов с системной красной волчанкой.
Для повышения информативности гистологического исследования биоптат должен быть получен из мышцы, соответствующей одному или более из следующих признаков:
слабость при клиническом исследовании,
Наличие признаков отека мышц на МРТ,
наличие изменений при электромиографии в контралатеральной парной мышце
Лабораторное исследование позволяет подкрепить или, напротив, снизить подозрение на наличие заболевания, оценить его тяжесть, выявить перекрестные изменения и осложнения. Показано определение аутоантител. Частота выявления антинуклеарных антител (АНА) у пациентов с дерматомиозитом и полимиозитом достигает 80%. Если ANA-тест положительный, то при усиливающемся подозрении в отношении перекрёстного синдрома важно проведение дальнейших исследований на наличие специфических типов антител.
Клиническое течение и проявления связаны с определенными антителами, как описано в таблицу аутоантител при аутоиммунном миозите. Связь между этими антителами и патогенезом заболевания пока неясна, хотя антитела к Jo-1 являются достоверным маркером фиброзирующего альвеолита, легочного фиброза, артрита и синдрома Рейно. Антител, специфичных для полимиозита не существует.
Данные относительно повышенного риска развития рака являются относительно достоверными для дерматомиозита и менее достоверными для полимиозита. Поэтому следует рассматривать вопрос скрининга на рак у пациентов ≥ 40 лет с дерматомиозитом или у пациентов ≥ 60 лет с полимиозитом, потому что у таких пациентов часто неожиданно выявляют рак. Скрининг должен включать по крайней мере объективное обследование, которое включает в себя: обследование молочных желез, тазовых органов и прямой кишки (с анализом на скрытую кровь); общий анализ крови; биохимический профиль; маммографию; анализ мочи; рентгенограмму органов грудной клетки; и любые другие тесты, соответствующие возрасту пациента.
Дополнительное исследование проводят с учетом данных анамнеза и физикального обследования. Некоторые руководства рекомендуют КТ грудной клетки, брюшной полости и таза, а также колоноскопию, особенно пациентам с дерматомиозитом. Пациентам более молодого возраста, не имеющим клинических признаков злокачественных опухолей, такой скрининг не требуется.
Прогноз при аутоиммунном миозите
Длительная ремиссия (даже клиническое выздоровление) в течение 5 лет отмечается у более чем половины прошедших лечение пациентов; у детей данный показатель выше. Рецидив тем не менее может развиться в любое время. Общая 5-летняя выживаемость составляет 75%, у детей она выше.
Летальному исходу у взрослых предшествуют выраженная и прогрессирующая мышечная слабость, дисфагия, снижение питания, аспирационная пневмония или дыхательная недостаточность, осложнившаяся легочной инфекцией.
Смерть детей с дерматомиозитом может наступить в результате кишечного васкулита.
Дерматомиозит и полимиозит связаны с повышенным риском развития рака. Общий прогноз заболевания определяется также наличием злокачественных новообразований.
Лечение аутоиммунного миозита
Кортикостероиды
Иммунодепрессанты (например, метотрексат, азатиоприн, микофенолата мофетил, ритуксимаб, такролимус)
Внутривенный иммуноглобулин (ВВИГ)
Двигательный режим должен быть щадящим до подавления воспаления.
Глюкокортикоиды являются средством выбора для начала терапии. При остром заболевании взрослые получают преднизон перорально в дозе 1 мг/кг день (обычно 40-60 мг) перорально 1 раз в день. При тяжелом заболевании с дисфагией или слабостью дыхательных мышц лечение обычно начинают с терапии высокими дозами кортикостероидов (например, метилпреднизолон от 0,5 до 1 г внутривенно 1 раз в день в течение 3–5 дней).
Регулярное определение активности креатинкиназы (КФК) является лучшим ранним показателем эффективности. Тем не менее при выраженной атрофии мышц активность фермента может быть нормальной несмотря на наличие хронического активного миозита. Данные МРТ мышечного отека или высокие значения активности креатинкиназы обычно помогают дифференцировать рецидив миозита и миопатию, индуцированную глюкокортикоидами. Альтернативой является определение уровня альдолазы, этот показатель менее специфичен при поражении мышц, чем КK, но может быть положительным у пациентов с миозитом и нормальными уровнями креатинкиназы. Поскольку у многих пациентов через 6–12 недель уровни ферментов падают или достигают нормальных значений, а затем улучшается мышечная сила, дозу кортикостероидов можно постепенно снижать. Если уровни мышечных ферментов снова повышаются, то дозу кортикостероидов обычно увеличивают, ожидая полного эффекта от других медикаментов.
Общая цель состоит в том, чтобы быстро исключить воспаление, но минимизировать воздействие длительного приема кортикостероидов, поэтому прием второго препарата (обычно метотрексата, такролимуса или азатиоприна в качестве нестероидных препаратов первой линии) начинают одновременно с кортикостероидами или вскоре после них, так, чтобы свести дозу преднизона до максимальной дозы 5 мг/день, идеально в течение примерно 6 месяцев. В/в иммунный глобулин является хорошим вариантом для пациентов, которые не реагируют быстро на лечение, у которых развиваются инфекционные осложнения при терапии высокими дозами кортикостероидов и другими иммунодепрессантами или пациентов, которые проходят химиотерапию. В тяжелых случаях или при наличии кортикостероидной интоксикации некоторые специалисты могут использовать комбинацию всех 3-х видов терапии. Начальная доза преднизона для детей составляет 30–60 мг/м2 день однократно.
В ряде случаев у пациентов, длительно получающих высокие дозы глюкокортикоидов, после первоначального ответа увеличивается слабость из-за развития стероидной безболевой миопатии. У таких пациентов уровень креатинкиназы остается нормальным, хотя пациенты являются более слабыми.
Миозит, ассоциированный с раком, обычно отличается большей рефрактерностью к кортикостероидам. Миозит, ассоциированный с раком, может стихать после ее удаления.
Пациенты с аутоиммунным заболеванием имеют повышенный риск развития атеросклероза и должны тщательно наблюдаться. При длительной терапии кортикостероидами пациентам следует проводить профилактику остеопороза. Если используется комбинированная иммуносупрессивная терапия, пациенты должны получать профилактику оппортунистических инфекций, таких как инфекция, вызванная Pneumocystis jirovecii (см. профилактику пневмонии, вызванной Pneumocystis jirovecii), и вакцинацию вакцинами против распространенных инфекций (например, стрептококковой пневмонии, гриппа, COVID-19).
Основные положения
Мышечная слабость, вызванная миозитом, чаще всего проксимальная.
Гелиотропная сыпь и папулы Готтрона специфичны для дерматомиозита.
Для уточнения диагноза следует искать характерную сыпь, мышечную слабость, повышение уровня креатинкиназы, а также изменения мышечных волокон на электромиографии или МРТ.
Если у пациентов нет характерных кожных высыпаний, для подтверждения диагноза выполняется биопсия мышечной ткани.
Следует рассмотреть возможность проведения онкоскрининга пациентов ≥ 40 лет с дерматомиозитом и пациентов ≥ 60 лет с полимиозитом.
Лечение пациентов проводится с применением кортикостероидов и других иммуносупрессоров.







