




A hemocromatose hereditária é uma doença genética caracterizada pelo acúmulo excessivo de ferro (Fe) que resulta em lesão tecidual. As manifestações podem incluir sintomas constitucionais, distúrbios hepáticos, cardiomiopatia, diabetes, disfunção erétil e artropatia. O diagnóstico é feito pelos altos níveis séricos de ferritina, ferro e saturação da transferrina, sendo confirmado por um exame genético. O tratamento é realizado por meio de sangrias terapêuticas seriadas.
(Ver também Visão geral da sobrecarga de ferro.)
Etiologia da hemocromatose hereditária
Existem 4 tipos de hemocromatose hereditária, tipos 1 a 4, dependendo do gene que sofre a mutação.
Tipo 1: mutações no gene HFE (regulador homeostático do ferro humano)
Tipo 2 (hemocromatose juvenil): mutações nos genes HJV (co-receptor BMP hemojuvelino) e HAMP (peptídeo antimicrobiano hepcidina)
Tipo 3: mutações no gene TFR2 (receptor de transferrina 2)
Tipo 4 (doença por ferroportina): mutações no gene SLC40A1 (família transportadora de soluto 40, membro 1)
Outras alterações genéticas muito mais raras podem causar sobrecarga de ferro hepático, mas o quadro clínico geralmente é dominado pelos sinais e sintomas decorrentes da insuficiência de outros órgãos (p. ex., anemia na hipotransferrinemia ou atransferrinemia, ou alterações neurológicas na aceruloplasminemia).
Embora esses tipos variem acentuadamente em relação à idade de início, as consequências clínicas da sobrecarga de ferro são as mesmas em todos os tipos (1).
Hemocromatose hereditária tipo 1
O tipo 1 é a hemocromatose hereditária clássica, também denominada hemocromatose relacionada com HFE. Mais de 80% dos casos são provocados pela mutação homozigótica C282Y ou mutação heterozigota do composto C282Y/H63D. Mutações homozigóticas H63D raramente ocorrem e têm o mesmo fenótipo dos casos de mutações homozigóticas C282y. O distúrbio é autossômico recessivo, com frequência homozigota de 1:200 e frequência heterozigota de 1:8 no norte da Europa. As mutações C282Y e H63D são incomuns entre afrodescendentes e raras entre pessoas com ascendência asiática. Dos pacientes com as características clínicas da hemocromatose, 83% são homozigotos. No entanto, por razões desconhecidas, a fenotípica da doença (clínica) é muito menos comum do que prevista pela frequência do gene (isto é, muitas pessoas homozigotas não manifestam o distúrbio).
Hemocromatose hereditária tipo 2
A hemocromatose hereditária tipo 2 (hemocromatose juvenil) é uma doença autossômica recessiva rara causada por mutações no gene HJV que afetam a transcrição da proteína hemojuvelina ou as mutações no gene HAMP, que codifica diretamente a hepcidina. Aparece, com frequência, em adolescentes.
Hemocromatose hereditária tipo 3
Mutações no receptor 2 da transferrina (TFR2), um gene que codifica para uma proteína que parece controlar a saturação da transferrina, podem causar uma forma rara, autossômica recessiva, de hemocromatose.
Hemocromatose hereditária tipo 4
A hemocromatose hereditária tipo 4 (doença por ferroportina) ocorre predominantemente em descendentes do sul da Europa. Ela resulta de uma mutação autossômica dominante no gene SLC40A1 e afeta a capacidade da ferroportina de ligar a hepcidina.
Deficiência de transferrina e ceruloplasmina
Na deficiência de transferrina (hipotransferrinemia ou atransferrinemia), o ferro absorvido que entra no sistema portal como ferro não ligado à transferrina é depositado no fígado. Transferência subsequente para os locais de produção de eritrócitos é reduzida em razão da deficiência de transferrina.
Na deficiência de ceruloplasmina (aceruloplasminemia), a ausência de ferroxidase provoca conversão defeituosa do Fe2+ em Fe3+; essa conversão é necessária para a ligação à transferrina. O defeito na ligação da transferrina prejudica o movimento do ferro dos depósitos intracelulares ao transporte plasmático, resultando no acúmulo de ferro nos tecidos.
Referência sobre etiologia
1. Pietrangelo A: Hereditary hemochromatosis: Pathogenesis, diagnosis, and treatment. Gastroenterology 139:393–408, 2010.
Fisiopatologia da hemocromatose hereditária
O conteúdo total normal de ferro no corpo é cerca de 2,5 g em mulheres e 3,5 g em homens. Como os sintomas podem ser atrasados até que o acúmulo do ferro seja excessivo (p. ex., > 10 a 20 g), a hemocromatose talvez só seja reconhecida mais tarde na vida, embora seja uma anormalidade adquirida. Em mulheres, manifestações clínicas são incomuns antes da menopausa porque a perda do ferro na menstruação (e, às vezes, na gestação e no parto) tende a compensar esse acúmulo.
O mecanismo da sobrecarga de ferro tanto na hemocromatose HFE como não HFE é o aumento da absorção de ferro pelo trato gastrointestinal, levando à deposição crônica do ferro nos tecidos. A hepcidina, um peptídeo derivado do fígado, é o mecanismo de controle crítico de absorção do ferro. A hepcidina costuma sofrer modulação positiva quando os depósitos de ferro são elevados e, por meio do seu efeito inibitório sobre a ferroportina (que participa da absorção de ferro), ela impede a absorção e o armazenamento do excesso de ferro nas pessoas hígidas. A hemocromatose dos tipos 1 a 4 compartilham a mesma base patológica (p. ex., ausência de síntese ou atividade da hepcidina) e as principais características clínicas.
Em geral, a lesão tecidual parece ser resultado dos radicais livres reativos de hidroxila gerados quando a deposição de ferro nos tecidos catalisa sua formação. Outros mecanismos podem afetar órgãos em particular (p. ex., hiperpigmentação da pele pode resultar de aumento de melanina, assim como de acúmulo de ferro). No fígado, a peroxidação lipídica associada ao ferro induz a apoptose dos hepatócitos, que estimula a ativação das células de Kupffer e a liberação de citocinas pró-inflamatórias. Essas citocinas ativam as células estreladas hepáticas para produzir colágeno, resultando no acúmulo patológico de fibrose no fígado e risco de carcinoma hepatocelular.
Sinais e sintomas da hemocromatose hereditária
As consequências clínicas da sobrecarga de ferro são semelhantes, independente da etiologia e da fisiopatologia da sobrecarga.
Historicamente, os especialistas acreditavam que os sintomas não se desenvolviam até que ocorresse dano significativo nos órgãos. Todavia, as lesões nos órgãos são lentas e graduais, e com frequência no início ocorrem fadiga e sinais e sintomas sistêmicos inespecíficos. Por exemplo, a disfunção hepática pode se manifestar insidiosamente com fadiga, dor abdominal no hipocôndrio direito e hepatomegalia. As alterações laboratoriais da sobrecarga de ferro e da hepatite costumam preceder os sintomas.
Na hemocromatose hereditária tipo 1 (HFE), os sintomas estão relacionados aos órgãos com os maiores depósitos de ferro (ver tabela Manifestações comuns da hemocromatose hereditária). Em homens, os sintomas iniciais podem ser hipogonadismo e disfunção erétil causada pela deposição de ferro nas gônadas. Intolerância à glicose ou diabetes mellitus é outro sintoma inicial comum. Alguns pacientes apresentam hipotireoidismo.
A complicação mais comum é a doença hepática que pode progredir para cirrose; 20 a 30% dos pacientes com cirrose desenvolvem carcinoma hepatocelular. A doença hepática é a causa mais comum de morte. Enzimas hepáticas elevadas são uma das anormalidades laboratoriais mais comuns em pacientes com hemocromatose tipo 1.
Cardiomiopatia com insuficiência cardíaca é a 2ª complicação fatal mais frequente. A hiperpigmentação (diabetes brônzeo) e a porfiria cutânea tardia são comuns, bem como a artropatia sintomática nas mãos.
Na doença tipo 2, os sinais e sintomas incluem hepatomegalia progressiva e hipogonadismo hipogonadotrópico.
Na doença tipo 3, os sinais e sintomas são similares aos da hemocromatose hereditária tipo 1 (HFE).

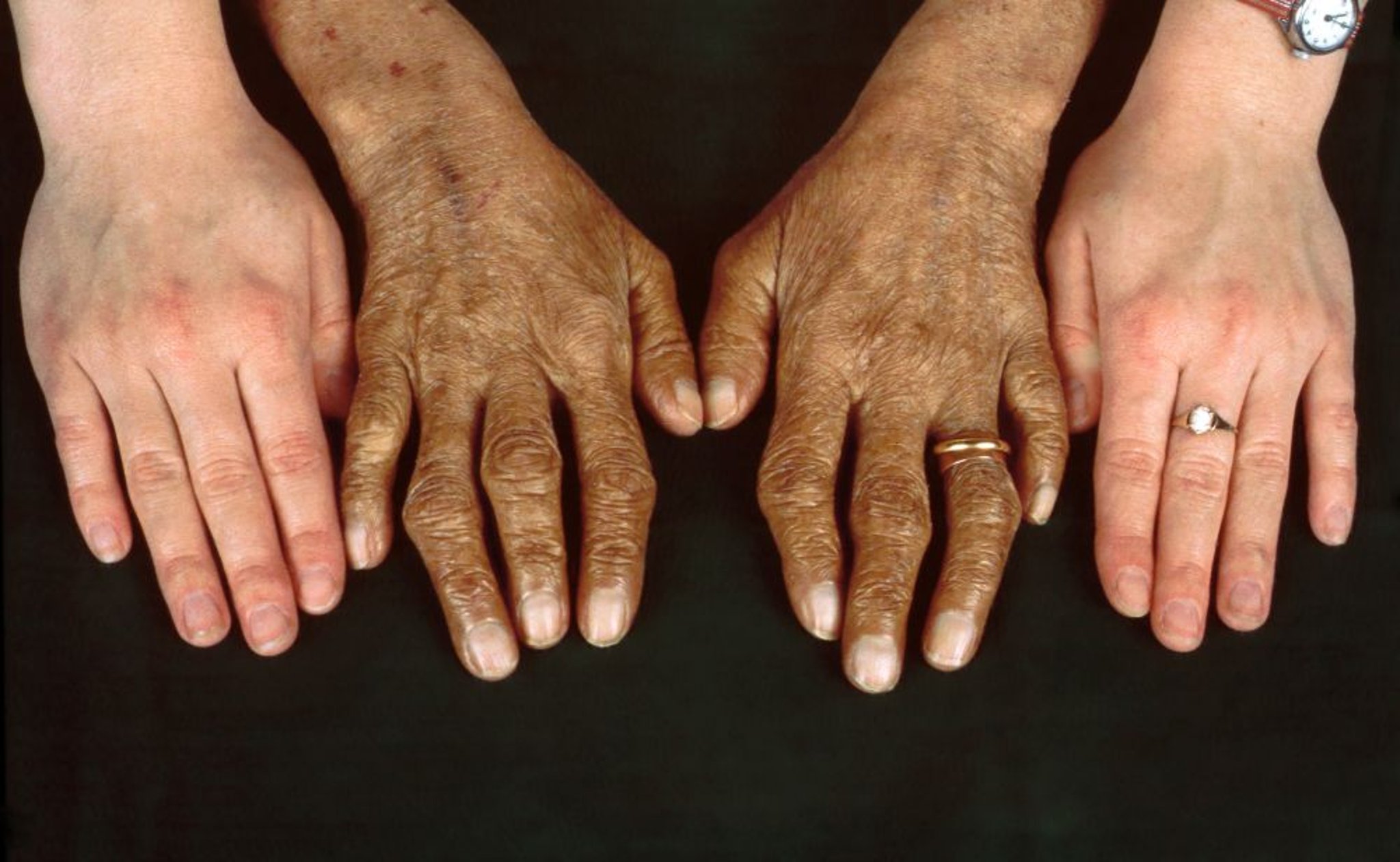
Clinical Photography/SCIENCE PHOTO LIBRARY
A doença tipo 4 manifesta-se na primeira década de vida como ferritina sérica aumentada com saturação de transferrina baixa ou normal; a saturação progressiva ocorre quando os pacientes estão na faixa de 20 e 30 anos. As manifestações clínicas são mais leves do que na doença tipo 1, com doença hepática modesta e anemia leve.
Manifestações comuns da hemocromatose hereditária
Ferritina sérica, ferro sérico de jejum e saturação da transferrina
Exame genético
Algumas vezes, biópsia hepática
Sinais e sintomas podem ser inespecíficos, sutis e de início gradual, para que haja alto índice de suspeita. Deve-se suspeitar de hemocromatose primária em pacientes com sintomas típicos, particularmente combinações desses sintomas, que permanecem sem explicação após a avaliação de rotina. História familiar de hemocromatose, cirrose ou carcinoma hepatocelular é um indício mais específico. Avaliar sobrecarga de ferro em todos os pacientes com hepatopatia crônica, independentemente de raça ou etnia
O nível de ferritina sérica é o teste inicial mais simples e mais direto. Níveis elevados (> 200 ng/mL [> 200 mcg/L] em mulheres ou > 250 ng/mL [> 250 mcg/L] em homens) normalmente estão presentes na hemocromatose hereditária, mas podem resultar de outras anomalias, como doenças hepáticas inflamatórias (p. ex., hepatite viral crônica, doença do fígado gorduroso não alcoólica, doença hepática alcoólica), câncer, determinadas doenças inflamatórias sistêmicas (p. ex., artrite reumatoide, linfo-histiocitose hemofagocítica) ou obesidade. Fazer exames adicionais se o nível de ferritina estiver alterado; esses exames são o ferro sérico de jejum (geralmente > 300 mg/dL [> 53,7 mcmol/L]) e a capacidade de fixação do ferro (saturação da transferrina; níveis geralmente > 50%). Uma saturação da transferrina < 45% tem valor preditivo negativo de 97% para a sobrecarga de ferro.
Na doença tipo 2, os níveis de ferritina são > 1.000 ng/mL (> 1000 mcg/L), a saturação da transferrina é > 90%.
Na deficiência de transferrina ou ceruloplasmina, os níveis séricos de transferrina ou ceruloplasmina (isto é, a capacidade de fixação do ferro) são extremamente baixos ou indetectáveis.
O teste do gene é um diagnóstico da hemocromatose hereditária causada por mutações do gene HFE. Cerca de 70% dos pacientes com mutações homozigóticas C282Y do gene HFE têm aumento dos níveis de ferritina, mas apenas cerca de 10% desses pacientes apresentam evidências de disfunção orgânica. A sobrecarga de ferro clinicamente significativa é ainda menos comum nos pacientes com mutações heterozigóticas do gene HFE (isto é, C282Y/H63D). Suspeita-se de hemocromatose tipos 2 a 4 nas instâncias menos comuns em que testes sanguíneos para ferro e ferritina indicam sobrecarga de ferro e o exame genético é negativo para mutação do gene HFE, particularmente em pacientes mais jovens. A confirmação desses diagnósticos por teste genético não está rotineiramente disponível.
Quando o diagnóstico é confirmado, deve-se examinar o fígado à procura de fibrose e cirrose. Até 80% dos pacientes com cirrose e uma mutação homozigótica C282Y terão ferritina > 1.000 ng/mL, aumento na AST (aspartato transaminase) e ALT (alanina transaminase) e contagem de plaquetas < 200 × 103/mcL (< 200 × 109/L). Como a cirrose modifica o prognóstico, quando a ferritina estiver > 1.000 ng/mL, costuma-se fazer biópsia hepática e dosar os níveis de ferro nos tecidos (quando disponível). Também recomenda-se biópsia hepática nos pacientes com evidências sorológicas de sobrecarga de ferro, porém com resultados negativos pela avaliação genética. RM com elastografia (RME) sem contraste é uma alternativa não invasiva para estimar a quantidade de ferro e de fibrose hepática, e está se tornando cada vez mais precisa.
É necessário fazer triagem dos parentes de primeiro grau de pessoas com hemocromatose hereditária dosando os níveis séricos de ferritina e testando as mutações C282Y e H63D no gene HFE.
Tratamento da hemocromatose hereditária
Flebotomia
O tratamento é indicado para pacientes com manifestações clínicas, níveis de ferritina sérica elevados (particularmente níveis > 1.000 ng/mL [> 1000 mcg/L]), ou saturação da transferrina elevada. Pacientes assintomáticos somente precisam de avaliação clínica periódica (p. ex., anual) e verificação dos níveis séricos de ferro, ferritina, saturação da transferrina e dosagem das enzimas hepáticas.
A sangria terapêutica é o método mais simples e mais eficaz para remoção do excesso de ferro. Prolonga a progressão de fibrose para cirrose, às vezes até reverte mudanças cirróticas e prolonga a sobrevida, mas não evita o carcinoma hepatocelular. Cerca de 500 mL de sangue (cerca de 250 mg de ferro) são removidos semanalmente ou a cada duas semanas até que os níveis séricos de ferritina alcancem 50 a 100 ng/mL. Pode ser necessário fazer flebotomia semanal ou a cada duas semanas durante muitos meses (p. ex., ao remover 250 mg de ferro por semana, serão necessárias 40 semanas para remover 10 g de ferro). Quando os níveis de ferro chegarem ao normal, as flebotomias podem ser intermitentes de modo a manter a ferritina entre 50 e 100 ng/mL.
Diabetes, cardiomiopatia, disfunção erétil e outras manifestações secundárias são tratadas conforme indicação. Deve-se fazer triagem de carcinoma hepatocelular para os pacientes com fibrose avançada ou cirrose por sobrecarga de ferro a cada 6 meses por meio de ultrassonografia hepática.
Os pacientes devem seguir dieta balanceada; não é necessário restringir o consumo de alimentos que contêm ferro (p. ex., carne vermelha, fígado). O álcool deve ser consumido apenas com moderação porque ele pode aumentar a absorção de ferro e, em altas doses, eleva o risco de cirrose. Deve-se evitar suplementos de vitamina C porque eles aumentam a absorção de ferro no duodeno.
Nos pacientes com doença tipo 4, a tolerância à flebotomia vigorosa é baixa; é necessário o monitoramento seriado do nível de hemoglobina e saturação de transferrina.
O tratamento da deficiência de transferrina e ceruloplasmina é experimental; p. ex., queladores de ferro podem ser mais bem tolerados do que a flebotomia porque os pacientes normalmente têm anemia.
Pontos-chave
Há 4 tipos de hemocromatose hereditária, todos os quais envolvendo mutações que prejudicam a capacidade do corpo em inibir a absorção de ferro quando as reservas de ferro são excessivas.
Os efeitos da sobrecarga de ferro são semelhantes em todos os tipos e incluem doença hepática (que leva à cirrose), pigmentação da pele, diabetes, artropatia, hipogonadismo e, algumas vezes, insuficiência cardíaca.
Diagnosticar dosando o nível sérico da ferritina; se elevado, confirmar demonstrando os níveis séricos de ferro, saturação da transferrina e exame genético.
Uma vez feito o diagnóstico, deve-se avaliar o grau de fibrose hepática por biópsia do fígado ou exames de imagem não invasivos, como elastografia por RM, para determinar o prognóstico; considerar exame genético e triagem de parentes de primeiro grau.
Tratar com flebotomia e moderação do consumo de álcool.







