




Amiloidose é qualquer de um grupo de condições distintas caracterizadas por deposição extracelular de fibrilas insolúveis compostas de proteínas mal agregadas. Essas proteínas podem se acumular localmente, causando relativamente poucos sintomas, ou se disseminar, envolvendo múltiplos órgãos e provocando graves insuficiências. A amiloidose pode ocorrer de novo ou ser secundária a várias doenças infecciosas, inflamatórias ou malignas. O diagnóstico é feito por biópsia do tecido afetado; a proteína amiloidogênica é tipificada utilizando uma variedade de técnicas imuno-histológicas e bioquímicas. O tratamento varia de acordo com o tipo de amiloidose.
Fibrilas amiloides são constituídas de proteínas mal dobradas com solubilidade normal que se agregam em oligômeros e logo se convertem em fibrilas insolúveis. Algumas proteínas normais (tipo selvagem) e mutantes são susceptíveis a esse entrelaçamento e agregação incorreto (proteínas amiloidogênicas), sendo assim responsáveis por uma grande variedade de causas e tipos de amiloidose.
Depósitos amiloides são compostas por pequenas fibrilas insolúveis (com cerca de 10 nm de diâmetro) que formam folhas congofílicas beta-plissadas que podem ser identificadas por difração de raios-x. Além da proteína fibrilar amiloide, os depósitos também contêm componente P de amiloide sérico e glicosaminoglicanos.
Depósitos amiloides têm coloração rosa com hematoxilina e eosina, contêm componentes de carboidratos que provocam coloração com corante periódico de ácido de Schiff ou azul de Alcian, mas a maioria tem caracteristicamente birrefringência verde-maçã sob microscopia de luz polarizada após coloração com vermelho Congo. Na inspeção da autópsia, os órgãos afetados podem ter aspecto seroso.
Para que a amiloidose se desenvolva, além da produção de proteínas amiloidogênicas, há também uma falha dos mecanismos normais de depuração para essas proteínas mal entrelaçadas. Os próprios depósitos amiloides são metabolicamente inertes, mas interferem fisicamente na estrutura e função dos órgãos. Mas alguns oligômeros pré-fibrilares das proteínas amiloidogênicas têm toxicidade celular direta, um componente importante da patogênese da doença.
Etiologia da amiloidose
Na amiloidose sistêmica, proteínas amiloidogênicas na circulação formam depósitos em uma variedade de órgãos. Os principais tipos sistêmicos incluem
AL (amiloidose primária): causada por superexpressão adquirida de cadeias leves de imunoglobulina clonal
AF (amiloidose familiar): causada por hereditariedade de um gene mutante que codifica uma proteína propensa a enovelamento incorreto, mais comumente transtirretina (TTR)
ATTRts (ATTR do tipo selvagem; anteriormente denominada amiloidose sistêmica senil ou ASS): causada por dobramento incorreto de proteínas com acúmulo de TTR do tipo selvavem
AA (amiloidose secundária): causada pela agregação de um reagente de fase aguda, amiloide A sérico
A amiloidose causada pela agregação da beta-2-microglobulina pode ocorrer em pacientes em hemodiálise há muito tempo, mas sua incidência diminuiu com a utilização das modernas membranas de diálise de alto fluxo. Há uma forma hereditária rara da amiloidose beta-2-microglobulina decorrente de uma mutação no gene relevante.
As formas localizadas da amiloidose parecem ser causadas pela produção local e depósito de uma proteína amiloidogênica (mais frequentemente imunoglobulinas de cadeias leves) no interior do órgão comprometido, em vez de por deposição de proteínas circulantes. Os locais que costumam ser atingidos são sistema nervoso central (p. ex., na doença de Alzheimer), pele, vias respiratórias superiores ou inferiores, parênquima pulmonar, bexiga, olhos e mamas.
Amiloidose AL (amiloidose primária)
AL é causada por superprodução de uma cadeia leve de imunoglobulina amiloidogênica em pacientes com células plasmáticas monoclonais ou outra doença linfoproliferativa de células B. As cadeias leves também podem formar depósitos de tecido não fibrilar (isto é, doença de deposição de cadeia leve). Raramente, as imunoglobulinas de cadeias pesadas formam fibrilas amiloides (chamada amiloidose AH).
Os locais comuns de deposição amiloide são pele, nervos, coração, trato gastrointestinal (inclusive a língua), rim, fígado, baço e vasos sanguíneos. Normalmente, plasmocitose leve está presente na medula óssea, que é semelhante a mieloma múltiplo, embora a maioria dos pacientes não tenha mieloma múltiplo verdadeiro (com lesões ósseas líticas, hipercalcemia, cilindros tubulares renais e anemia). Entretanto, cerca de 10 a 20% dos pacientes com mieloma múltiplo desenvolvem amiloidose AL.
Amiloidose AF (amiloidose familiar)
AF é causada por herança de um gene que codifica uma proteína sérica mutada propensa à agregação, geralmente uma proteína abundantemente produzida pelo fígado.
As proteínas séricas que causam AF são transtirretina (TTR) apolipoproteína A-I e apolipoproteína A-II, lisozima, fibrinogênio, gelsolina e cistatina C. Uma forma em que especulas-se que pode ser familiar é causada pelo fator quimiotático 2 de leucócitos na proteína sérica (LECT2); entretanto, uma mutação genética herdada específica para esse último tipo não foi claramente demonstrada.
Amiloidose causada por TTR (ATTR) é o tipo mais comum da AF. Mais de 130 mutações do gene TTR foram associadas à amiloidose. A mutação mais prevalente, V30M, é comum em Portugal, Suécia, Brasil e Japão, e a mutação V122I está presente em cerca de 4% dos negros norte-americanos e caribenhos. A penetrância e idade no início da doença são altamente variáveis, mas são consistentes com famílias e grupos étnicos (1).
A ATTR causa neuropatia sensório-motora e neuropatia autonômica, doença renal crônica e cardiomiopatia. A síndrome do túnel do carpo geralmente precede outras manifestações de doença neurológica. Os depósitos vítreos ocorrer pela produção de TTR mutante no epitélio retiniano, ou pode haver depósitos leptomeníngeos se o plexo coroide produzir TTR mutante. Quando a miocardiopatia é a manifestação predominante da deposição de TTR no coração, ela é chamada de miocardiopatia amiloide por transtirretina (ATTR-CM).
Amiloidose ATTRts (amiloidose sistêmica senil)
ATTRts é causada por agregação e depósito de TTR do tipo selvagem, principalmente direcionado ao coração.
ATTRts é cada vez mais reconhecida como causa de cardiomiopatia infiltrativa em homens idosos. Aproximadamente 16% dos pacientes com estenose aórtica submetidos à substituição valvar aórtica transcateter (2) e 13% daqueles hospitalizados por insuficiência cardíaca com fração de ejeção preservada (ICFEp) também têm cardiomiopatia amiloide transtiretina, nesse caso designada como wATTR-CM para denotar deposição de TTR de tipo selvagem no coração (3). Manifestações nos tecidos moles da amiloide ATTRwt, incluindo síndrome do túnel do carpo, ruptura do tendão bicipital, ruptura do manguito rotador e estenose espinal, podem preceder a expressão clínica da cardiomiopatia infiltrativa em anos.
Os fatores genéticos e epigenéticos que levam à ATTRts são desconhecidos. Como a amiloidose ATTRts e AL podem causar cardiomiopatia, e como gamopatias monoclonais amiloidogênicas podem se identificadas em pacientes nessa faixa etária, é essencial fazer a tipagem exata da amiloide para que os pacientes com ATTRts não sejam tradados inadequadamente com quimioterapia (indicada para a AL).
Amiloidose AA (amiloidose secundária)
Essa forma pode ocorrer em várias condições infecciosas, inflamatórias e doenças malignas e é causada pela agregação de isoformas do reagente de fase aguda amiloide sérico A.
As infecções causadoras comuns incluem
As doenças inflamatórias predisponentes são
Síndromes de febre periódica herdadas, como febre familiar do mediterrâneo
Doença de Castleman
As citocinas inflamatórias [p. ex., interleucina (IL)-1, fator de necrose tumoral (TNF), IL-6] que são produzidas nessas doenças ou de forma ectópica pelas células tumorais causam aumento da síntese hepática da amiloide A sérica (SAA).
Amiloidose AA mostra predileção pelos rins, baço, fígado, glândulas suprarrenais e linfonodos. O comprometimento do coração o dos nervos periféricos e autonômicos ocorre tardiamente no curso da doença.
Amiloidose localizada
A amiloidose localizada fora do encéfalo é mais frequentemente causada por depósitos de imunoglobulina clonal de cadeias leves; no interior do encéfalo, predomina a proteína beta-amiloide.
Depósitos amiloides localizados tipicamente envolvem as vias respiratórias e tecido pulmonar, bexiga e ureteres, pele, mamas e olhos. Raramente, outras proteínas produzidas localmente causam amiloidose, como queratina isoforme que pode formar depósitos localmente na pele. Cadeias leves de imunoglobulina clonal produzidas pelo tecido linfoide associado à mucosa no trato gastrointestinal, vias respíratórias e bexiga podem resultar em AL localizada nesses órgãos.
Os depósitos de proteína beta-amiloide no cérebro contribuem para a doença de Alzheimer ou a angiopatia amiloide cerebrovascular. Outras proteínas produzidas no sistema nervoso central pode ter mal formação, agregação e danos nos neurônios, levando a doenças neurodegenerativas (p. ex., doença de Parkinson, doença de Huntington).
Referências sobre etiologia
1. Buxbaum JN, Ruberg FL: Transthyretin V122I (pV142I)* cardiac amyloidosis: an age-dependent autosomal dominant cardiomyopathy too common to be overlooked as a cause of significant heart disease in elderly African Americans. Genet Med 19(7):733-742, 2017. doi:10.1038/gim.2016.200
2. Fabbri G, Serenelli M, Cantone A, et al: Transthyretin amyloidosis in aortic stenosis: clinical and therapeutic implications. Eur Heart J Suppl 23(Suppl E):E128-E132, 2021. doi:10.1093/eurheartj/suab107
3. Magdi M, Mostafa MR, Abusnina W, et al: A systematic review and meta-analysis of the prevalence of transthyretin amyloidosis in heart failure with preserved ejection fraction. Am J Cardiovasc Dis 12(3):102-111, 2022. PMID: 35873185
Sinais e sintomas da amiloidose
Os sinais e sintomas da amiloidose sistêmica são inespecíficos, muitas vezes resultando em atrasos no diagnóstico. Deve-se suspeitar mais fortemente de amiloidose em pacientes com um processo de doença multissistêmica progressiva.
Depósitos amiloides renais tipicamente ocorrem na membrana glomerular levando à proteinúria, mas em cerca de 15% dos casos, os túbulos são afetados, causando azotemia com proteinúria mínima. Esses processos podem evoluir para síndrome nefrótica com hipoalbuminemia acentuada, edema e anasarca ou para a fase final de doença renal.
O envolvimento hepático causa hepatomegalia indolor, que pode ser maciça. Testes hepáticos geralmente sugerem colestase intra-hepática com elevação da fosfatase alcalina e, mais tarde, bilirrubina, embora icterícia seja rara. Ocasionalmente, desenvolve-se hipertensão portal, resultando em varizes de esôfago e ascite.
O envolvimento das vias respiratórias e laríngeas resulta em dispneia, rouquidão, sibilos, hemoptise ou obstrução das vias respiratórias.
Infiltração do miocárdio causa cardiomiopatia restritiva, levando com o tempo à disfunção diastólica e insuficiência cardíaca; bloqueio cardíaco ou arritmia pode ocorrer. Hipotensão é comum.
A neuropatia periférica é uma manifestação clínica comum em todas as amiloidoses primárias (AL) e familiares (ATTR), com parestesias nos dedos das mãos e dos pés. A neuropatia autonômica pode causar hipotensão ortostática, disfunção erétil, anormalidades de sudorese, retenção urinária e distúrbios de motilidade gastrointestinal.
A angiopatia amiloide cerebrovascular mais frequentemente causa hemorragia cerebral espontânea, mas alguns pacientes têm sintomas neurológicos transitórios breves.
O amiloide gastrointestinal pode causar anormalidades de motilidade do esôfago e dos intestinos grosso e delgado. Atonia gástrica, má absorção, sangramento ou pseudo-obstrução também podem ocorrer. Macroglossia é comum na amiloidose AL.
Caracteristicamente, o envolvimento amiloide dos tecidos moles precede a expressão clínica da cardiomiopatia amiloide da ATTRts. Manifestações da doença amiloide das partes moles incluem síndrome do túnel do carpo, dedo em gatilho, ruptura do tendão bicipital e estenose espinal.

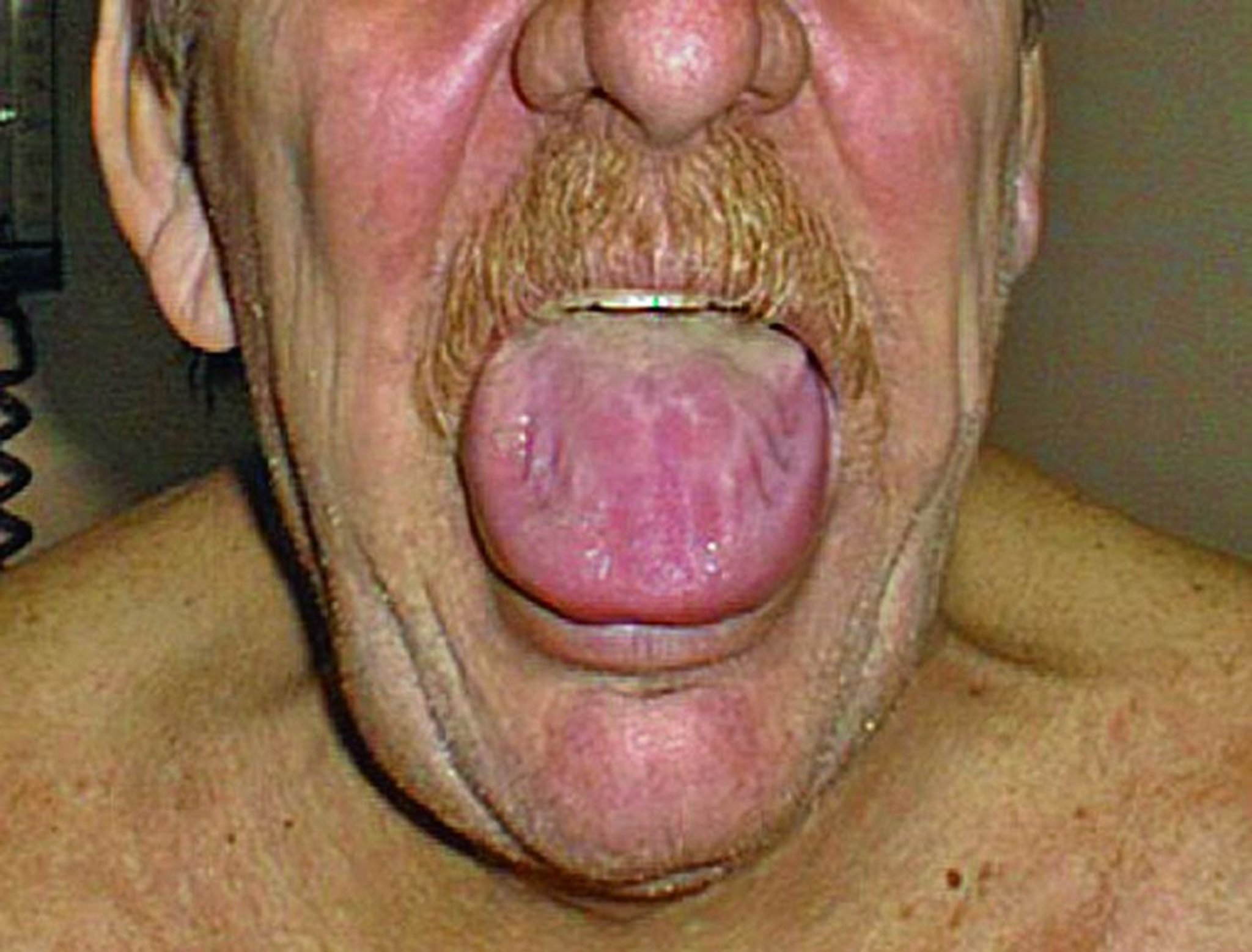
© Springer Science+Business Media
A amiloidose da glândula tireoide pode causar bócio simétrico, de consistência firme e indolor que se assemelha ao encontrado na tireoidite de Hashimoto. Outras endocrinopatias também podem ocorrer.
O envolvimento pulmonar (sobretudo na amiloidose AL) pode ser caracterizado por nódulos e cistos pulmonares focais, lesões traqueobrônquicas, derrames pleurais ou depósitos alveolares-septais (intersticiais) difusos.
Ocorre opacidades vítreas amiloides e margens pupilares entrecortada bilaterais em várias amiloidoses hereditárias.
Outras manifestações incluem hematomas, com frequência em volta dos olhos (olhos de guaxinim), causados por depósitos amiloides nos vasos sanguíneos. Os depósitos amiloides enfraquecem os vasos sanguíneos, que podem se romper após pequenos traumas, como espirros ou tosse.
Diagnóstico da amiloidose
Biópsia
Tipagem amiloide
Teste para envolvimento de órgãos
Biópsia
O diagnóstico da amiloidose é feito pela demonstração de depósitos fibrilares em um órgão envolvido. A aspiração da gordura abdominal subcutânea detecta depósitos amiloides em cerca de 80% dos pacientes com AL mas em menos de 25% dos pacientes com ATTRts (1). Se o resultado da biópsia de gordura é negativo, um órgão clinicamente envolvido deve passar por biópsia. A sensibilidade diagnóstica das biópsias renais e cardíacas é quase 100% quando esses órgãos estão clinicamente comprometidos. As seções teciduais são coradas com vermelho Congo e examinadas em microscópio com luz polarizada para verificar a birrefringência característica. Fibrilas de 10 nm sem ramificações também podem ser reconhecidas por microscopia eletrônica em amostras de biópsia de coração ou rim.
A cintilografia utilizando marcadores ósseos pode diagnosticar a cardiomiopatia amiloide ATTR sem biópsia cardíaca, desde que a amiloidose AL seja descartada.
Tipagem amiloide
Depois de a amiloidose ser confirmada por biópsia, o tipo é determinado utilizando uma variedade de técnicas. Para alguns tipos de amiloidose, imuno-histoquímica ou imunofluorescência pode ser diagnóstica, mas ocorrem resultados falso-positivos de tipagem. Outras técnicas úteis são o sequenciamento de genes para AF e a identificação bioquímica por espectrometria de massa identificar com precisão as variantes proteicas nos depósitos amiloides (o método mais sensível e específico).
Se houve suspeita de AL, deve-se avaliar nos pacientes doença subjacente de células plasmáticas utilizando uma medição quantitativa das cadeias leves livres de imunoglobulina no plasma, detecção qualitativa de cadeias leves monoclonais no plasma ou na urina utilizando eletroforese de imunofixação (eletroforese de proteínas séricas e eletroforese de proteínas na urina são insensíveis em pacientes com AL), e uma biópsia da medula óssea com citometria de fluxo ou imuno-histoquímica para estabelecer a clonalidade das células plasmáticas.
Deve-se testar se os pacientes com > 10% de células plasmáticas clonais para ver se correspondem aos critérios de mieloma múltiplo, com triagem de lesões ósseas líticas, anemia, insuficiência renal e hipercalcemia.
Envolvimento de órgãos
Os pacientes fazem o rastreamento de comprometimento de órgãos começando com os exames não invasivos:
Rins: exame de urina; dosagem de ureia, creatinina e albumina; taxa de filtração glomerular estimada (TFGe); e coleta de urina de 24 horas para eletroforese de proteínas (EP)
Fígado: provas de função hepática
Pulmão: radiografia de tórax, TC do tórax e provas de função pulmonar
Coração: ECG e mensuração dos biomarcadores como peptídeo natriurético encefálico (PNE) (tipo B) ou pró-PNE N-terminal (pro-PNE-NT) e troponina
O envolvimento cardíaco pode ser sugerido por baixa tensão no ECG (causada por ventrículo espessado) e/ou arritmias. Se houver suspeita de envolvimento cardíaco devido aos sintomas, além dos achados no ECG e dos biomarcadores cardíacos, realiza-se uma ecocardiografia para medir o relaxamento diastólico e a deformação longitudinal global (uma medida da função sistólica ventricular esquerda) e para rastrear hipertrofia biventricular. Em casos ambíguos, pode-se realizar RM cardíaca para detectar realce subendocárdico persistente pelo gadolínio, um achado característico. A cintilografia cardíaca com pirofosfato marcado com tecnécio melhora a detecção da amiloidose ATTR e podem evitar a necessidade de biópsias cardíacas, desde que os exames de sangue excluam a amiloidose AL (2, 3).
Referências sobre diagnóstico
1. Aimo A, Emdin M, Musetti V, et al: Abdominal Fat Biopsy for the Diagnosis of Cardiac Amyloidosis. JACC Case Rep 2(8):1182-1185, 2020. doi:10.1016/j.jaccas.2020.05.062
2. Gillmore JD, Maurer MS, Falk RH, et al: Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation 133(24):2404–2412, 2016.
3. Maurer MS, Bokhari S, Damy T, et al: Expert consensus recommendations for the suspicion and diagnosis of transthyretin cardiac amyloidosis. Circ Heart Fail 12(9):e006075, 2019.
Tratamento da amiloidose
Cuidados de suporte
Tratamento para tipo específico
Existem tratamentos específicos para a maioria das formas de amiloidose, embora algumas terapias estejam sob investigação. Para todas as formas da amiloidose sistêmica, medidas de tratamento de suporte podem ajudar a aliviar os sintomas e melhorar a qualidade de vida.
Cuidados de suporte
Medidas de tratamento de suporte são direcionadas para o sistema do órgão afetado:
Renal: pacientes com síndrome nefrótica e edema devem ser tratados com restrição de sal e líquidos e diuréticos de alça; por causa da perda de proteínas contínua, a ingestão de proteínas não deve ser restringida. O transplante renal é uma opção quando o processo da doença subjacente está controlado, e pode fornecer sobrevida a longo prazo comparável àquela de outras doenças renais.
Cardíaco: pacientes com cardiomiopatia devem ser tratados com restrição de sal e líquidos e diuréticos de alça. Outros fármacos para insuficiência cardíaca, como digoxina, inibidores da enzima conversora da angiotensina (ECA), bloqueadores dos canais de cálcio, e betabloqueadores, são mal tolerados e contraindicados. O transplante de coração foi bem-sucedido em pacientes cuidadosamente selecionados com amiloidose AL ou ATTR e comprometimento cardíaco grave. Para prevenir a recorrência no coração transplantado, os pacientes com amiloidose AL devem receber quimioterapia agressiva direcionada ao distúrbio clonal plasmocitário, e pacientes com polineuropatia amiloidótica sintomática ATTR ou cardiomiopatia devem ser considerados para fazer tratamento anti-TTR.
Gastrointestinal: pacientes com diarreia podem se beneficiar da loperamida. Aqueles com saciedade precoce e retenção gástrica podem se beneficiar da metoclopramida.
Sistema nervoso: nos pacientes com neuropatia periférica, a gabapentina ou a pregabalina podem aliviar a dor.
A hipotensão ortostática frequentemente melhora com doses elevadas de midodrina; esse fármaco pode causar retenção urinária em homens idosos, mas a complicação medicamentosa da hipertensão em decúbito dorsal raramente é um problema nessa população. Meias de suporte também podem ajudar, e fludrocortisona pode ser utilizada em pacientes sem edema periférico, anasarca ou insuficiência cardíaca. Para pacientes com hipotensão ortostática refratária, pode-se administrar midodrina, fludrocortisona ou droxidopa.
Amiloidose AL
Para amiloidose AL:
Início imediato da terapia celular antiplasmática é essencial para preservar a função do órgão e prolongar a vida.
A maioria dos fármacos utilizados para o mieloma múltiplo tem sido utilizada na amiloidose AL; a escolha do fármaco, a dose e a posologia muitas vezes devem ser modificados quando o órgão está funcionalmente prejudicado.
Quimioterapia com um agente de alquilação (p. ex., melfalano, ciclofosfamida) combinado com corticoides foi o primeiro regime a mostrar algum benefício. Altas doses de melfalano venoso, combinado com transplante autólogo de células-tronco, pode ser muito eficaz para alguns pacientes (1).
Inibidores de proteassoma (p. ex., bortezomibe) e imunomoduladores (p. ex., lenalidomida) também podem ser eficazes. Um ensaio clínico do anticorpo monoclonal daratumumabe mais ciclofosfamida, bortezomibe e dexametasona em pacientes com amiloidose AL recém-diagnosticada [excluindo aqueles com insuficiência cardíaca classe III e IV da NYHA, proteína natriurética N-terminal do tipo pro-B (NTproBNP) > 8.500 pg/mL (> 1005 pmol/L) e TFGe < 20 mL/minuto/m2] mostraram uma taxa alta sem precedentes de resposta hematológica (2). A resposta hematológica baseia-se nos níveis séricos de proteína monoclonal, nos níveis urinários determinados por eletroforese de imunofixação, e nos níveis séricos de cadeias leves com proporções capa/lambda. No entanto, não há dados de sobrevida de longo prazo.
Todos os tratamentos disponíveis têm como alvo células B clonais ou plasmócitos na amiloidose AL. Estudos de anticorpos antifibrilas, como birtamimabe e CAEL-101, estão em andamento (3).
A amiloidose AL localizada pode ser tratada com radioterapia de baixa dose com feixe externo porque as células plasmáticas são altamente radiossensíveis.
Amiloidose ATTR
Para amiloidose ATTR:
Transplante de fígado
Fármacos estabilizadores de tetrâmero
Fármacos de silenciamento de genes
Transplante de fígado — que substitui o local primário da síntese hepática de proteína mutante por um novo órgão que produz TTR normal — pode ser eficaz em certas mutações TTR se feito no início da doença (neuropatia em fase precoce e sem comprometimento cardíaco). Transplante mais tarde no curso da doença muitas vezes leva à cardiomiopatia e neuropatia amiloide progressiva devido ao enovelamento incorreto e à deposição de proteína TTR do tipo selvagem nos depósitos amiloides preexistentes.
Vários fármacos mostraram estabilizar tetrâmeros TTR circulando no plasma, inibindo o enovelamento incorreto do TTR e a formação de fibrilas e desacelerando de modo eficaz a progressão da doença neurológica e, ao mesmo tempo, preservando a qualidade de vida. Esses estabilizadores de TTR são o diflunisal, um anti-inflamatório amplamente disponível e o tafamidis. (4, 5).
O silenciamento do gene TTR utilizando RNA antissenso ou interferência de RNA para bloquear a tradução do TTR mRNA reduz eficazmente os níveis séricos de TTR, melhora os resultados neurológicos em cerca de 50% dos pacientes e, em alguns pacientes, parece capaz de reparar os nervos lesionados (6, 7). Medicamentos de silenciamento genético, como patisiran, inotersen e vutrisiran, estão disponíveis.
Um estudo com vutrisiran, um silenciador genético de segunda geração, demonstrou melhores resultados funcionais em pacientes com polineuropatia amiloide familiar (8). Dados preliminares de outro estudo sugerem que silenciadores genéticos podem ser eficazes no tratamento da cardiomiopatia em pacientes com amiloidose ATTR (9).
Amiloidose ATTRts
Para amiloidose ATTRts:
Fármacos estabilizadores de tetrâmero
A estabilização da TTR utilizando tafamidis, em pacientes com cardiomiopatia amiloide ATTR ou ATTRts demonstrou diminuir a mortalidade por todas as causas e as hospitalizações cardiovasculares (5). Estão em andamento ensaios clínicos que examinam o efeito dos silenciadores do gene TTR na cardiomiopatia que ocorre em pacientes com amiloidose ATTRwt, bem como na cardiomiopatia que ocorre em pacientes com amiloidose ATTR caracterizada pela proteína mutante (10).
Diferentemente da amiloidose ATTR hereditária, o transplante hepático não é eficaz para pacientes com ATTRts porque a proteína amiloidogênica é uma TTR estruturalmente normal.
Amiloidose AA
Para a amiloidose AA causada por febre familiar do Mediterrâneo, colchicina por via oral é eficaz.
Para outros tipos de AA, o tratamento é direcionado à doença infecciosa ou inflamatória ou ao câncer subjacente.
Pode-se colchicina e utilizar fármacos anti-IL1, anti-IL6 e anti-TNF para interromper a sinalização de citocinas, diminuindo o processo inflamatório que leva à produção hepática de proteína amiloide A sérica (SAA).
Referências sobre o tratamento
1. Sanchorawala V, Sun F, Quillen K, et al: Long-term outcome of patients with AL amyloidosis treated with high-dose melphalan and stem cell transplantation: 20-year experience. Blood 126: 2345–2347, 2015. doi: 10.1182/blood-2015-08-662726
2. Kastritis E, Palladini G, Minnema MC, et al: Daratumumab-Based Treatment for Immunoglobulin Light-Chain Amyloidosis. N Engl J Med 385(1):46-58, 2021. doi:10.1056/NEJMoa2028631
3. Quarta CC, Fontana M, Damy T, et al: Changing paradigm in the treatment of amyloidosis: From disease-modifying drugs to anti-fibril therapy. Front Cardiovasc Med 9:1073503, 2022. doi:10.3389/fcvm.2022.1073503
4. Berk JL, Suhr OB, Obici L, et al: Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA 310: 2658–2667, 2013. doi: 10.1001/jama.2013.283815
5. Maurer MS, Schwartz JH, Gundapaneni B, et al: Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med 379:1007–1016, 2018.
6. Adams D, Gonzalez-Duarte A, O'Riordan WD, et al: Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med 379:11–21, 2018.
7. Benson MD, Waddington-Cruz M, Berk JL, et al: Inotersen treatment for patients with transthyretin amyloidosis. N Engl J Med 379:22–31, 2018.
8. Adams D, Tournev IL, Taylor MS, et al: Efficacy and safety of vutrisiran for patients with hereditary transthyretin-mediated amyloidosis with polyneuropathy: a randomized clinical trial. Amyloid 30(1):1-9, 2023. doi:10.1080/13506129.2022.2091985
9. Maurer MS, Fontanta MA, Berk JL, et al: Primary results from APOLLO-B, a phase 3 study of patisiran in patients with transthyretin-mediated amyloidosis with cardiomyopathy. Abstract presented at International Symposium of Amyloidosis, September 2022,Heidelberg Germany.
10. Writing Committee, Kittleson MM, Ruberg FL, et al: 2023 ACC Expert Consensus Decision Pathway on Comprehensive Multidisciplinary Care for the Patient With Cardiac Amyloidosis: A Report of the American College of Cardiology Solution Set Oversight Committee [published correction appears in J Am Coll Cardiol 81(11):1135, 2023]. J Am Coll Cardiol 81(11):1076-1126, 2023. doi:10.1016/j.jacc.2022.11.022
Prognóstico para amiloidose
O prognóstico depende do tipo de amiloidose e sistema orgânico envolvido, mas com tratamento de suporte apropriado específico para a doença, muitos pacientes têm excelente expectativa de vida.
AL complicada por cardiomiopatia grave tem o prognóstico reservado, com sobrevida mediana de < 1 ano. A amiloidose ATTR não tratada geralmente evolui para doença cardíaca ou neurológica de estágio terminal em 5 a 15 anos. Anteriormente considerava-se que a ATTRts tinha a progressão mais lenta de todas as amiloidoses sistêmicas comprometendo o coração; mas alguns pacientes com ATTRts evoluem para insuficiência cardíaca sintomática e morte em uma mediana de 4 anos a partir do diagnóstico da biópsia.
O prognóstico na amiloidose AA depende em grande parte da eficácia do tratamento da doença infecciosa, inflamatória ou maligna subjacente.
Pontos-chave
A amiloidose é um grupo de doenças em que certas proteínas com mal formação se agregam em fibrilas insolúveis que são depositadas dentro dos órgãos, causando disfunção.
Muitas proteínas diferentes são propensas a mal formações; algumas dessas proteínas são produzidas por um defeito genético ou por certos estados de doença, enquanto outras envolvem cadeias leves de imunoglobulina produzidas por células monoclonais plasmáticas ou outras doenças linfoproliferativas de células-B.
A proteína amiloidogênica determina o tipo amiloide e o curso clínico da doença, embora as manifestações clínicas dos diferentes tipos possam se sobrepor.
Muitos órgãos podem ser afetados, mas o envolvimento cardíaco apresenta um prognóstico particularmente ruim; cardiomiopatia amiloide normalmente leva à disfunção diastólica, insuficiência cardíaca, bloqueio cardíaco ou arritmia.
O diagnóstico é por biópsia; o tipo de amiloidose é determinado por uma variedade de testes imunológicos, genéticos e bioquímicos. A espectrometria de massa é o método mais sensível e específico para a tipagem amiloide.
Tratamento de suporte apropriado ajudará a aliviar os sintomas e melhorar a qualidade de vida; transplante de órgão pode ajudar pacientes selecionados.
Tratar o processo subjacente; na amiloidose AL decorrente de células plasmáticas ou doenças linfoproliferativas, a quimioterapia pode ser muito eficaz; na amiloidose AA secundária, antimicrobianos e anti-inflamatórios podem ajudar.
Na amiloidose ATTR hereditária, terapias estabilizadoras com pequenas moléculas e fármacos de silenciamento de genes inibem ou potencialmente revertem a deterioração neurológica; para os pacientes com cardiomiopatia amiloide (ATTR ou ATTRts), tafamidis diminui a mortalidade por todas as causas e as hospitalizações cardiovasculares.







