




A sarcoidose é uma doença inflamatória que resulta em granulomas sem caseificação em um ou mais órgãos e tecidos; a etiologia é desconhecida. Os pulmões e o sistema linfático são afetados com mais frequência, mas a sarcoidose pode comprometer qualquer órgão. Os sintomas pulmonares variam de nenhum a tosse, dispneia durante esforço e, raramente, insuficiência respiratória ou de outro órgão. Primeiramente, de maneira geral, presume-se o diagnóstico pelo comprometimento pulmonar, confirmando-se por radiografia de tórax, biópsia e exclusão de outras causas de inflamação granulomatosa. O tratamento geralmente é indicado em pacientes sintomáticos. O tratamento de primeira linha é com corticoides. O prognóstico é excelente para a doença limitada, mas é ruim para a doença mais avançada.
A sarcoidose mais comumente afeta indivíduos na faixa etária de 20 a 40 anos, mas, eventualmente, acomete crianças e idosos. A prevalência mundial é maior em americanos de cor negra e norte-europeus étnicos, especialmente os escandinavos. A apresentação da doença varia amplamente de acordo com a raça e antecedentes étnicos. Pacientes norte-americanos negros desenvolvem manifestações extratorácicas com maior frequência. A sarcoidose é um pouco mais prevalente em mulheres.
Síndrome de Löfgren
A síndrome de Löfgren se manifesta como uma tríade de poliartrite aguda, eritema nodoso e adenopatia hilar. Febre, mal-estar, uveíte e parotidite também podem estar presentes. A síndrome de Löfgren é mais comum em pessoas de ascendência europeia. A síndrome de Löfgren é autolimitada. Os pacientes em geral podem ser tratados apenas com anti-inflamatórios não esteroides (AINEs). A taxa de recidiva é baixa.
Síndrome de Heerfordt
A síndrome de Heerfordt (febre uveoparotídea) se manifesta como edema da glândula parótida (devido à infiltração por sarcoide), uveíte, febre crônica e, menos frequentemente, paralisia do nervo facial. A síndrome de Heerfordt pode ser autolimitada. O tratamento é o mesmo que para sarcoidose.
Síndrome de Blau
A síndrome de Blau é uma doença semelhante à sarcoidose herdada de forma autossômica dominante que se manifesta em crianças. Não se sabe se a síndrome de Blau surge pelo mesmo mecanismo que a sarcoidose diagnosticada em adultos. Na síndrome de Blau, crianças apresentam antes dos 4 anos artrite, erupções cutâneas e uveíte. A síndrome de Blau geralmente é autolimitada. Os sintomas costumam ser alivados com AINEs.
Etiologia da sarcoidose
Admite-se que a sarcoidose decorra de resposta inflamatória exagerada a um antígeno ambiental em indivíduos geneticamente suscetíveis. Embora incerto, os deflagradores propostos incluem
Propionibacterium acnes e micobactérias (potencialmente a proteína catalase-peroxidase do Mycobacterium tuberculosis [mKatG])
Mofo ou bolor e certas substâncias não identificadas presentes nos locais de trabalho com odores de mofo
Pesticidas, sobretudo aqueles que contêm compostos de alumínio
O tabagismo está inversamente correlacionado com sarcoidose.
Evidências que apoiam a susceptibilidade genética incluem:
Taxa mais alta de concordância da doença em gêmeos monozigóticos do que gêmeos dizigóticos
Maior prevalência de sarcoidose (cerca de 3,6 a 9,6%) entre os parentes de 1ª ou 2ª grau de pacientes que têm sarcoidose
Aumento de cinco vezes no risco relativo de desenvolver sarcoidose em irmãos de pacientes que têm sarcoidose
Identificação de vários antígenos leucocitários humanos (HLA), e não HLA possivelmente associados ao risco, curso e fenótipos de sarcoidose
Por exemplo, o haplótipo HLA-DRB1*03/DQB1*02 está associado à síndrome de Löfgren e prediz um prognóstico excelente, em contraste com o HLA-DRB1*15/HLA DQB1*0602, que prediz uma doença persistente.
Fisiopatologia da sarcoidose
O antígeno desconhecido deflagra resposta imunitária mediada por célula, caracterizada pelo acúmulo de células T e macrófagos, liberação de citocinas e quimiocinas e organização de células responsivas em granulomas. Agrupamentos da doença em famílias e comunidades sugerem predisposição genética, exposições compartilhadas ou, com menos frequência, transmissão de pessoa a pessoa.
O resultado do processo inflamatório leva à formação de granulomas sem caseificação, a marca registrada da sarcoidose. Granulomas são coleções de células mononucleares e macrófagos, que se diferenciam em células epitelioides multinucleadas gigantes e são circundadas por linfócitos, plasmócitos, fibroblastos e colágeno. Granulomas ocorrem mais comumente nos pulmões e linfonodos, mas podem envolver qualquer órgão e causar disfunção significativa. Os granulomas no pulmão distribuem-se ao longo dos vasos linfáticos, encontrados predominantemente nas regiões peribronquiolar, subpleural e perilobular. O acúmulo de granulomas distorce a arquitetura dos órgãos afetados. Não se sabe se os granulomas levam diretamente à fibrose ou se seu curso é paralelo.
Pode ocorrer hipercalcemia por causa da maior conversão da vitamina D à forma ativada (1,25 di-hidroxi vitamina D) pelos macrófagos. Hipercalciúria pode estar presente, mesmo em pacientes com níveis séricos normais de cálcio sérico. Nefrolitíase e nefrocalcinose podem ocorrer, levando algumas vezes à doença renal crônica.

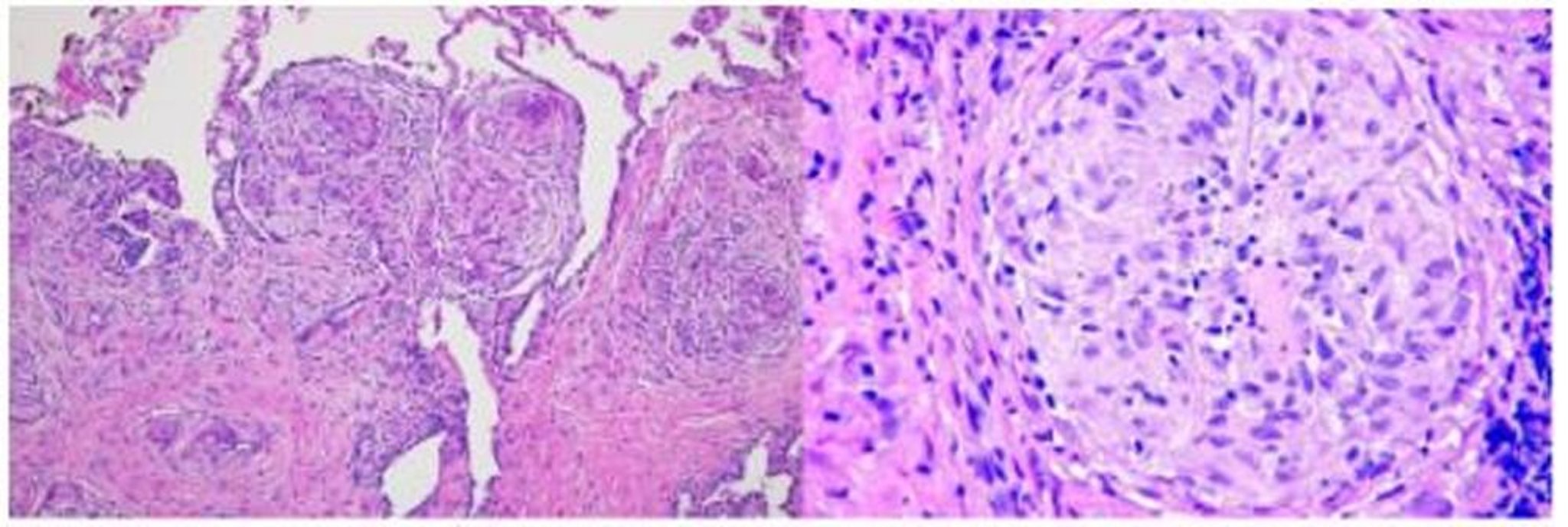
Image courtesy of Birendra P. Sah, MD, FCCP.
Sinais e sintomas da sarcoidose
Os sinais e sintomas dependem do local e do grau de comprometimento e variam no decorrer do tempo, variando da remissão espontânea à doença crônica indolente. Assim, é necessária a reavaliação frequente para a identificação de novos sintomas e envolvmento de diferentes órgãos. Provavelmente, a maioria dos casos é assintomática e, por isso, evolui sem ser detectada. Ocorre doença pulmonar em > 90% dos pacientes adultos.
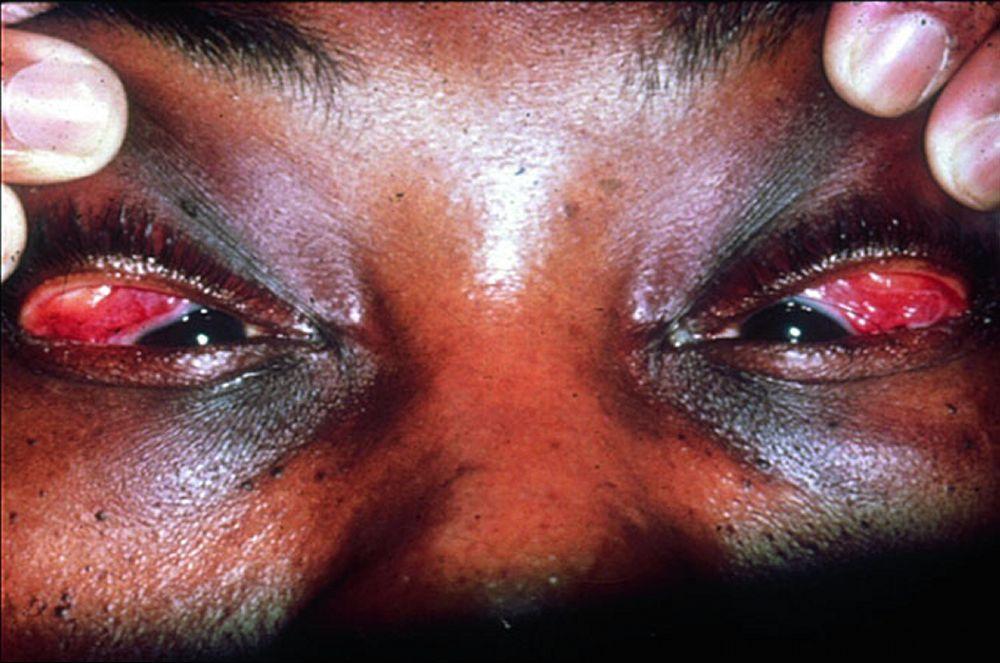
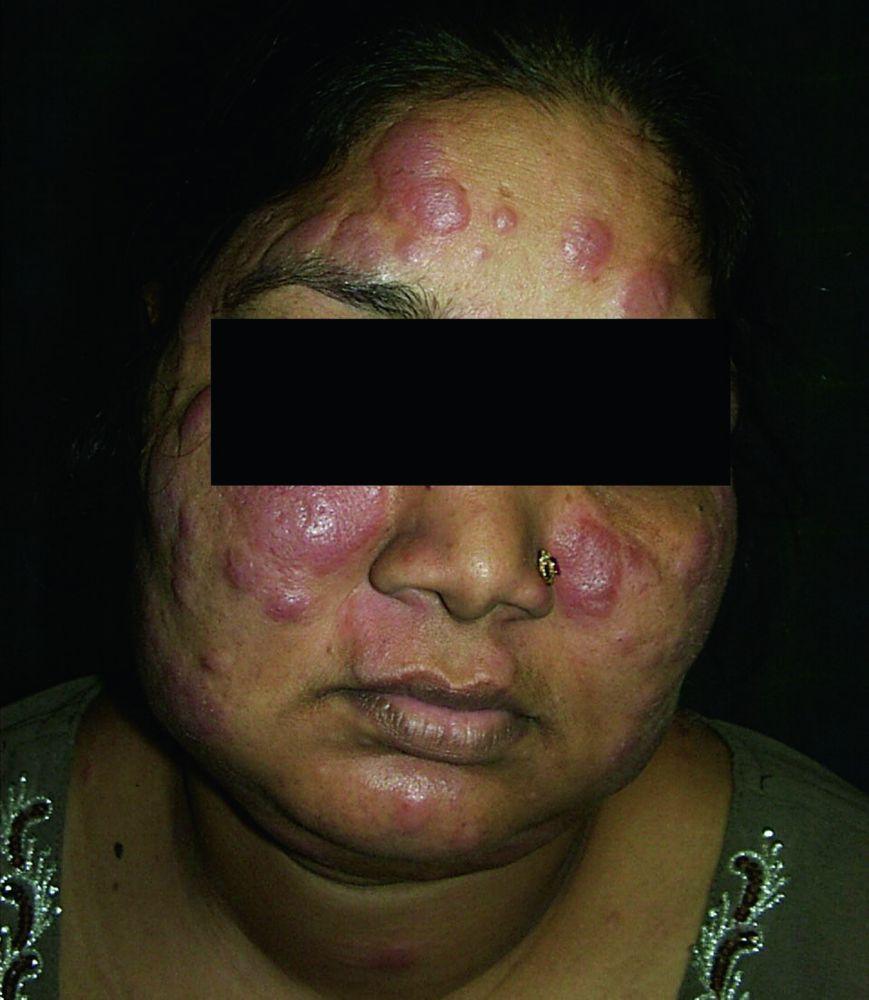
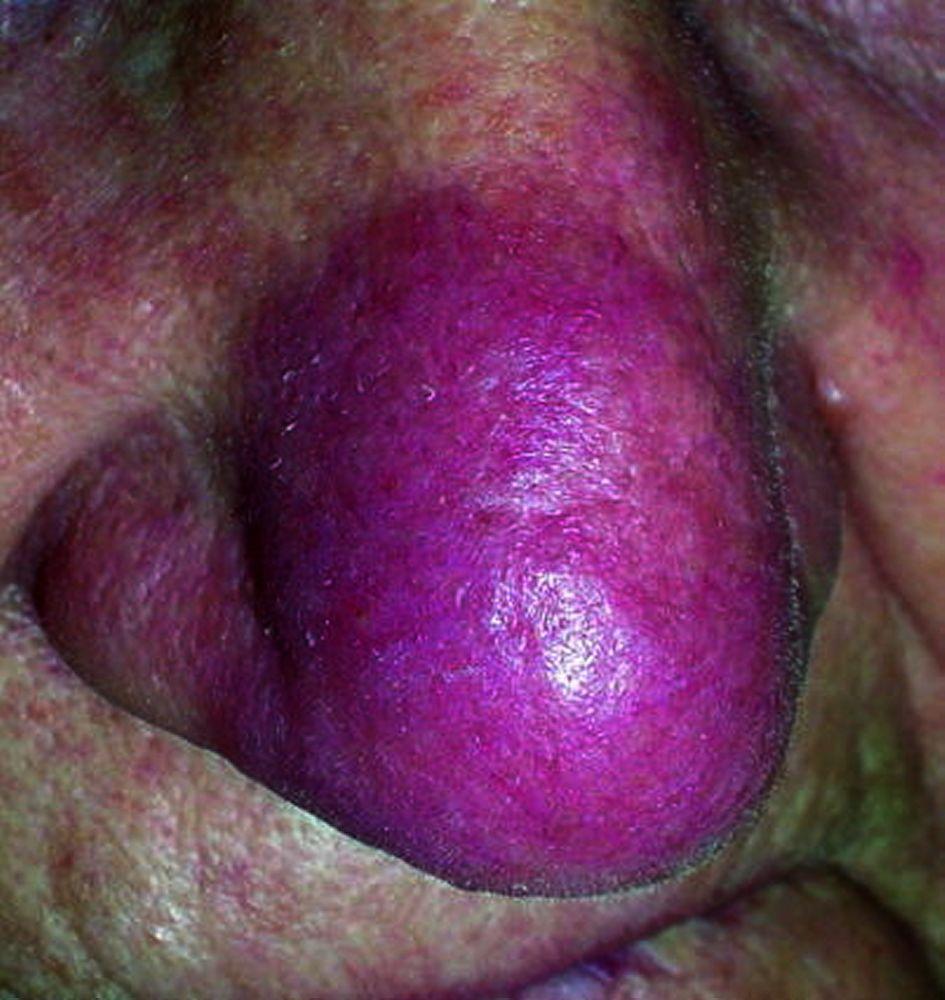
Os sinais e sintomas podem compreender dispneia, tosse, desconforto torácico e estertores crepitantes no exame físico. Também são comuns fadiga, mal-estar, fraqueza, anorexia, perda ponderal e febre baixa. A sarcoidose pode se manifestar como febre de origem indeterminada. O comprometimento sistêmico causa vários sintomas (ver tabela Comprometimento sistêmico na sarcoidose), os quais variam em termos de raça, sexo e idade. Os negros têm maior probabilidade que os brancos de desenvolverem comprometimento de olhos, fígado, medula óssea, linfonodos periféricos e pele, diferentemente do eritema nodoso. As mulheres têm maior probabilidade de desenvolver eritema nodoso e envolvimento dos olhos ou do sistema nervoso. Os pacientes do sexo masculino e os mais idosos têm maior probabilidade de ter hipercalcemia.
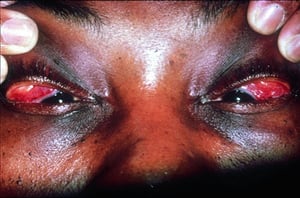
© Springer Science+Business Media
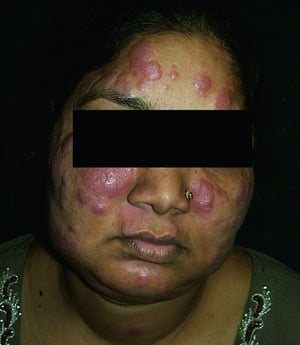
© Springer Science+Business Media
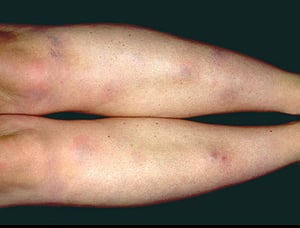
Image provided by Thomas Habif, MD.
© Springer Science+Business Media
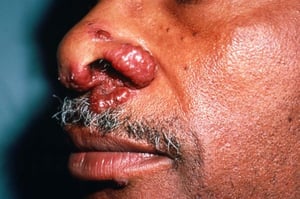
Image courtesy of Karen McKoy, MD.
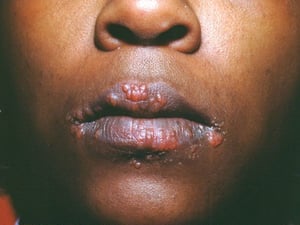
Image courtesy of Karen McKoy, MD.
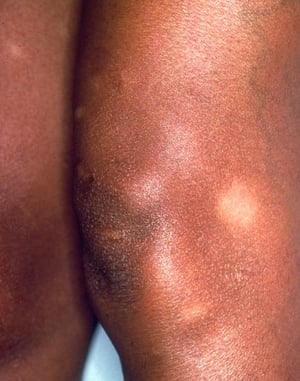
Image courtesy of Karen McKoy, MD.

© Springer Science+Business Media

© Springer Science+Business Media

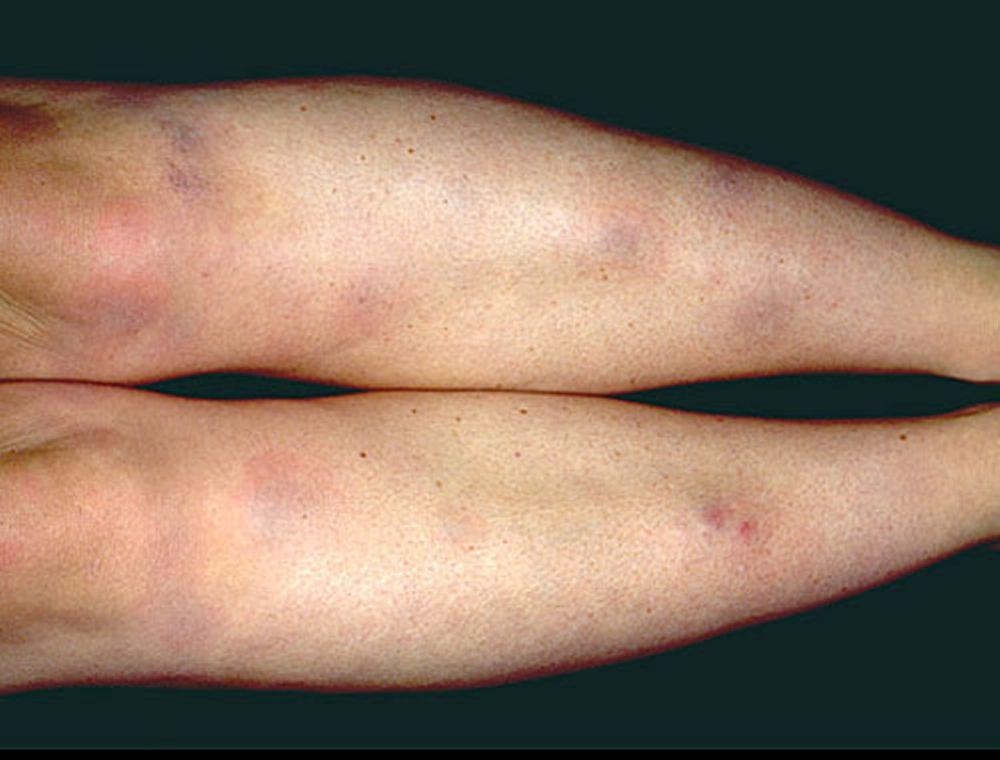
Image provided by Thomas Habif, MD.

© Springer Science+Business Media

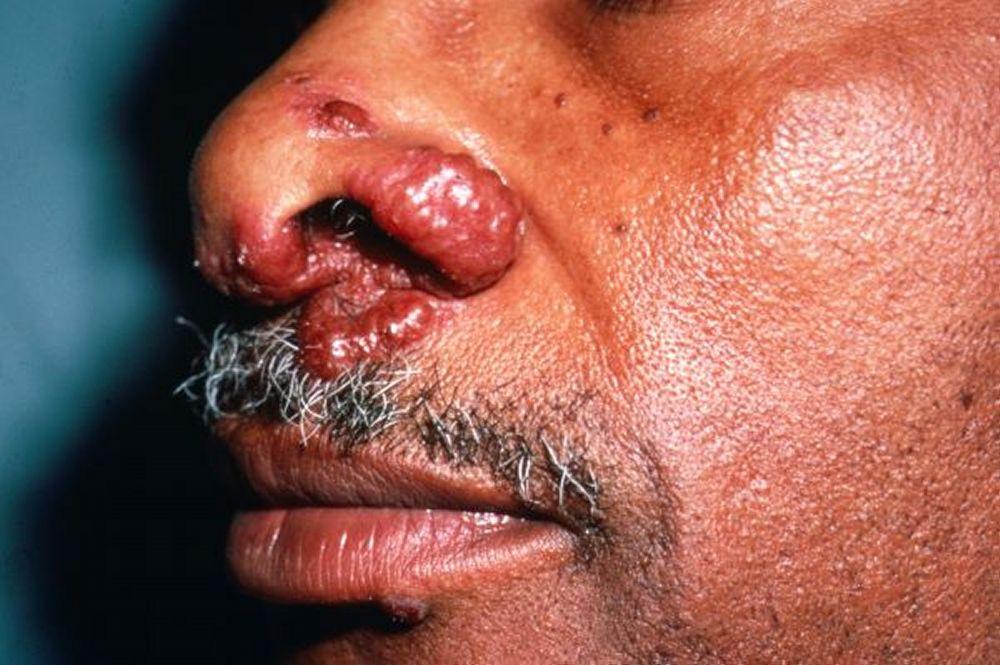
Image courtesy of Karen McKoy, MD.

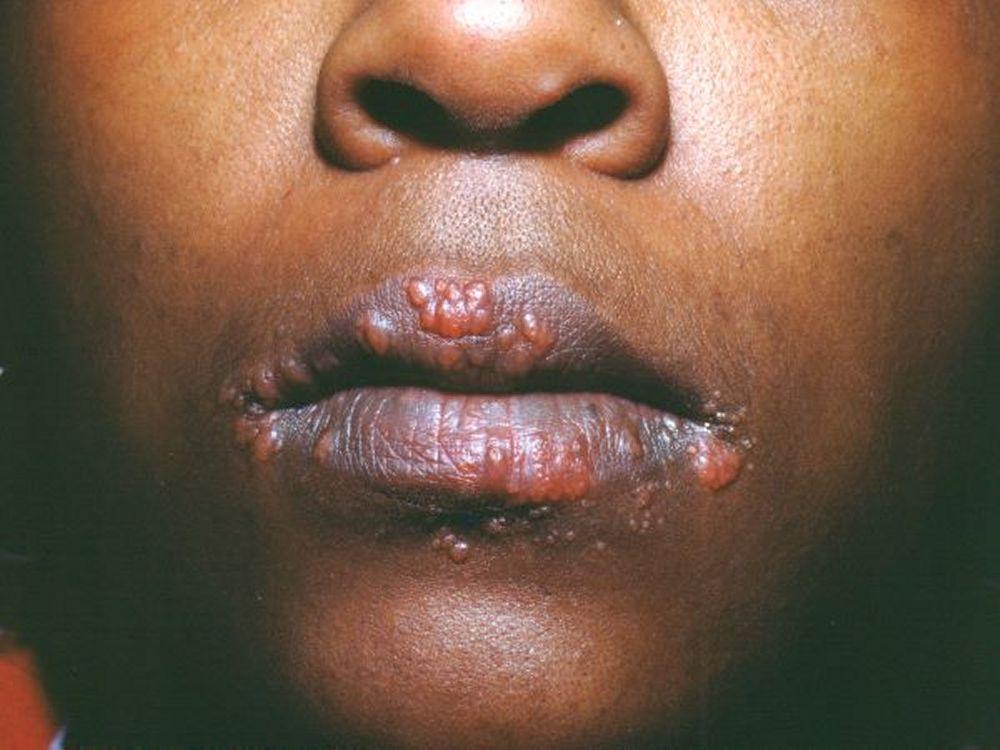
Image courtesy of Karen McKoy, MD.

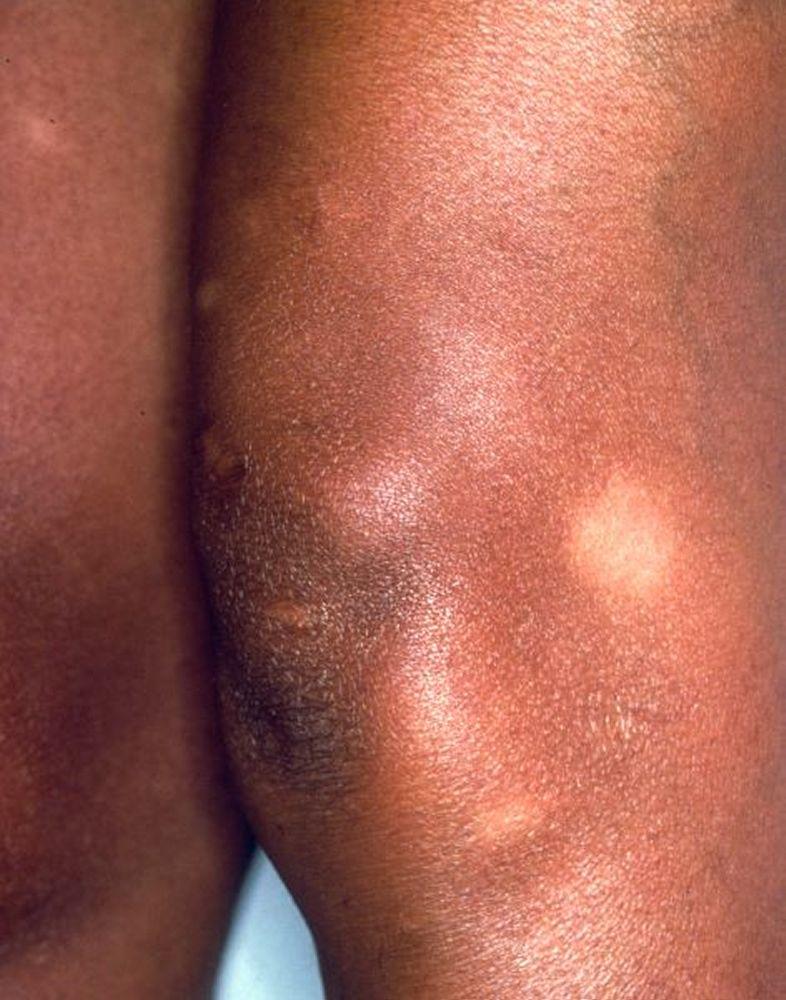
Image courtesy of Karen McKoy, MD.
Crianças com sarcoidose podem apresentar síndrome de Blau (artrite, exantema, uveíte) ou manifestações semelhantes às dos adultos. A sarcoidose pode ser confundida com artrite idiopática juvenil (artrite reumatoide juvenil) nessa faixa etária.
Diagnóstico da sarcoidose
Exames de imagem do tórax
Biópsia
Exclusão de outras doenças granulomatosas
Com maior frequência, a sarcoidose é presumida quando se detecta por acidente a linfonodopatia hilar, com ou sem infiltrados pulmonares, na radiografia de tórax. Adenopatia hilar bilateral é a anormalidade mais comum.
Se houver suspeita de sarcoidose, radiografia de tórax deve ser o primeiro exame se ainda não tiver sido feito. A aparência radiográfica tende a predizer aproximadamente a probabilidade da remissão espontânea (ver tabela Estadiamento da sarcoidose pela radiografia de tórax) nos pacientes com apenas comprometimento dos linfonodos torácicos. Contudo, o estadiamento da sarcoidose pela radiografia de tórax pode ser enganoso; por exemplo, a sarcoidose extrapulmonar, como a sarcoidose cardíaca ou neurológica, pode indicar um prognóstico desfavorável na ausência de comprometimento pulmonar. Além disso, os achados das radiografias de tórax não são bons preditores da função pulmonar, de modo que a aparência radiográfica do tórax pode não indicar com precisão a gravidade da sarcoidose pulmonar.
Radiografia de tórax normal (fase 0) não exclui o diagnóstico de sarcoidose, particularmente quando há suspeita de comprometimento cardíaco ou neurológico. TC de alta resolução é mais sensível para detectar linfadenopatia hilar e mediastinal e anormalidades parenquimais. O envolvimento do parênquima pulmonar ocorre predominantemente nos lobos superiores, mas pode ocorrer em qualquer parte dos pulmões. Achados da TC (ver também uma imagem de TC do tórax na sarcoidose pulmonar) nos estádios mais avançados (II a IV) incluem:
Espessamento dos feixes broncovasculares e das paredes brônquicas
Aspecto de contas dos septos interlobulares
Opacificação em vidro fosco
Nódulos parenquimatosos, cistos ou cavidades
Bronquiectasias por tração
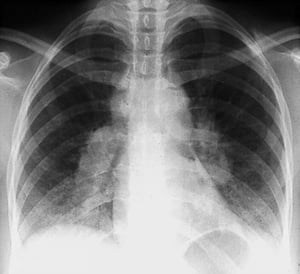
By permission of the publisher. De Tanoue L, Elias J. In Bone's Atlas of Pulmonary and Critical Care Medicine. Editado por J Crapo. Philadelphia, Current Medicine, 2005.
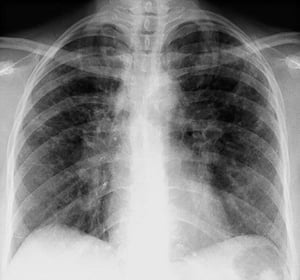
By permission of the publisher. De Tanoue L, Elias J. In Bone's Atlas of Pulmonary and Critical Care Medicine. Editado por J Crapo. Philadelphia, Current Medicine, 2005.
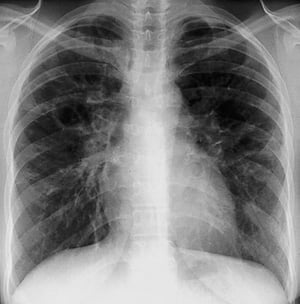
By permission of the publisher. De Tanoue L, Elias J. In Bone's Atlas of Pulmonary and Critical Care Medicine. Editado por J Crapo. Philadelphia, Current Medicine, 2005.
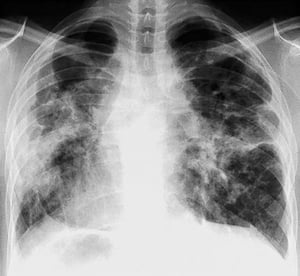
By permission of the publisher. De: Tanoue L, Elias J. In Bone's Atlas of Pulmonary and Critical Care Medicine. Editado por J Crapo. Philadelphia, Current Medicine, 2005.
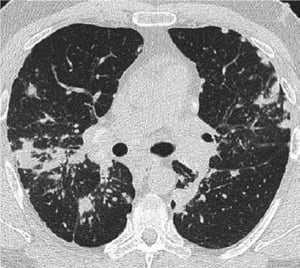
Image courtesy of Birendra P. Sah, MD, FCCP.

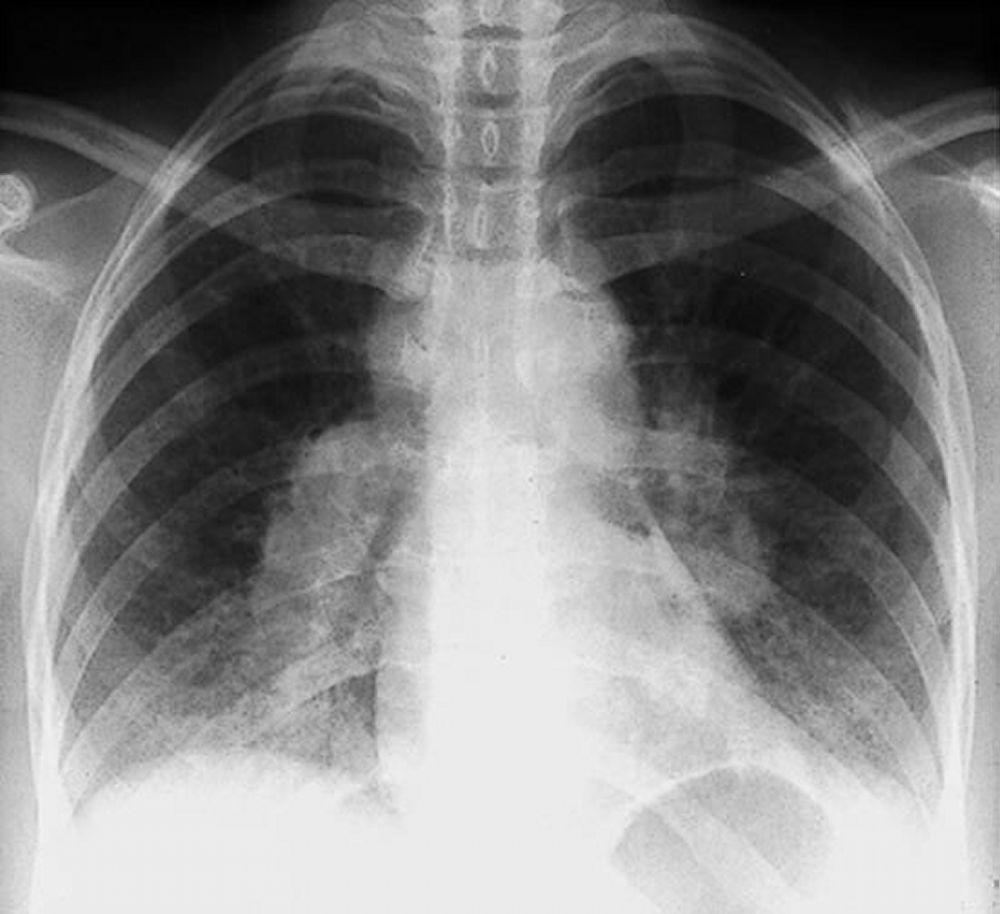
By permission of the publisher. De Tanoue L, Elias J. In Bone's Atlas of Pulmonary and Critical Care Medicine. Editado por J Crapo. Philadelphia, Current Medicine, 2005.

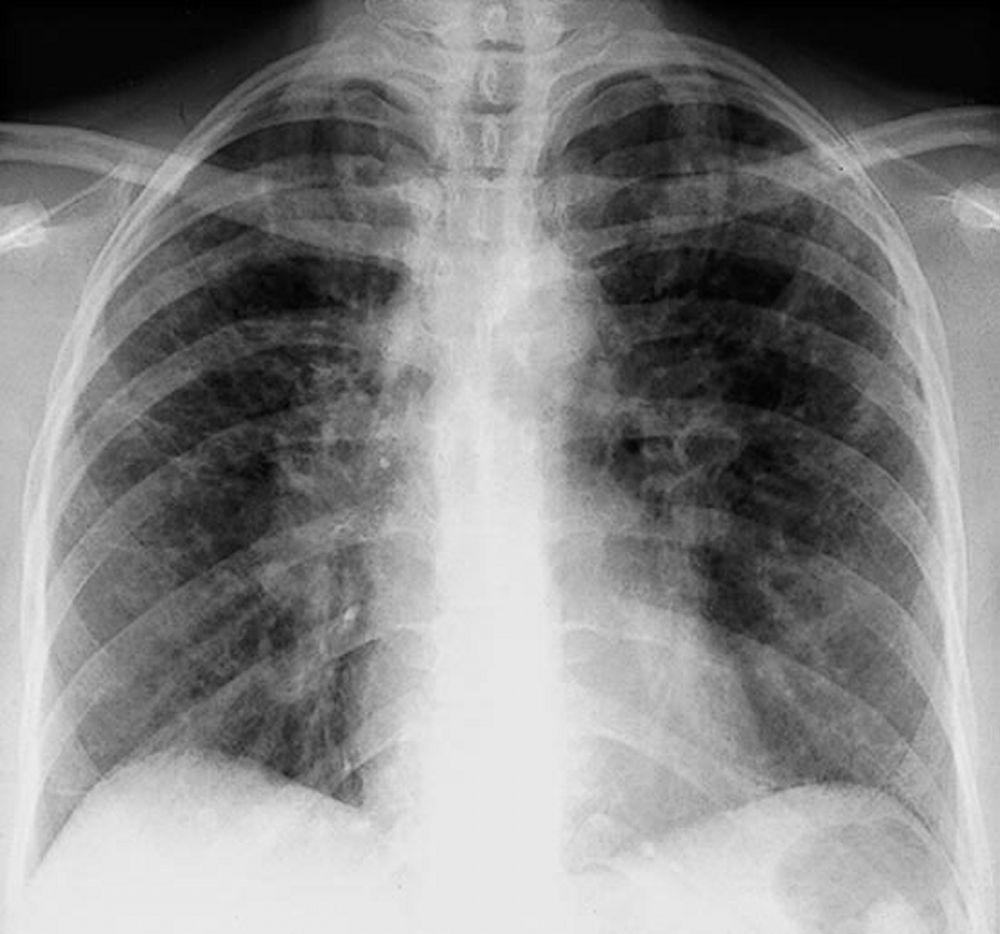
By permission of the publisher. De Tanoue L, Elias J. In Bone's Atlas of Pulmonary and Critical Care Medicine. Editado por J Crapo. Philadelphia, Current Medicine, 2005.

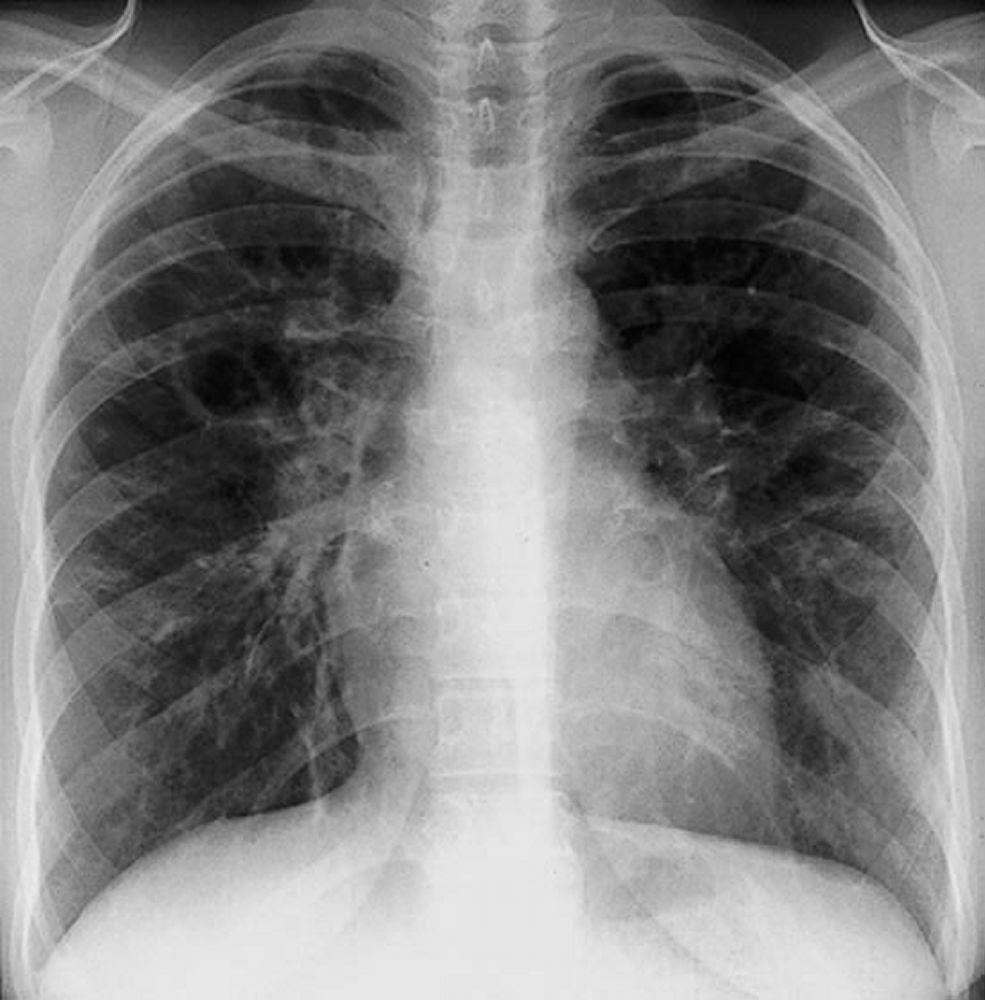
By permission of the publisher. De Tanoue L, Elias J. In Bone's Atlas of Pulmonary and Critical Care Medicine. Editado por J Crapo. Philadelphia, Current Medicine, 2005.

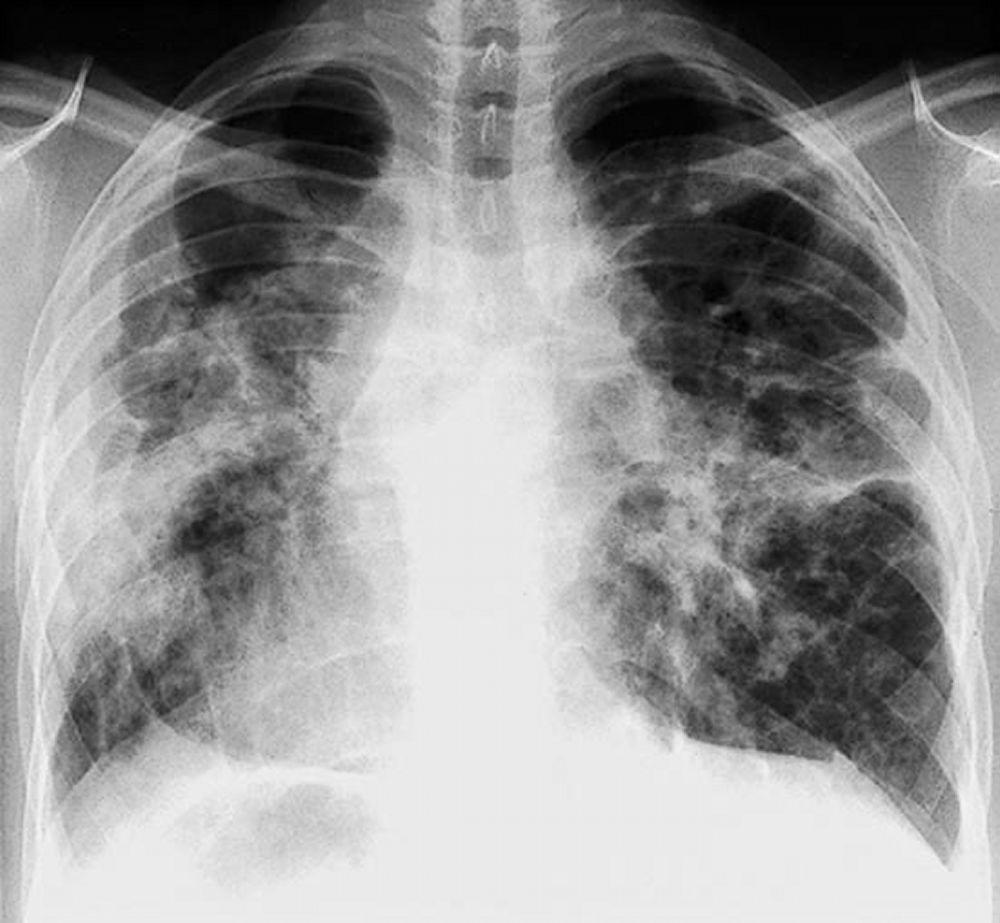
By permission of the publisher. De: Tanoue L, Elias J. In Bone's Atlas of Pulmonary and Critical Care Medicine. Editado por J Crapo. Philadelphia, Current Medicine, 2005.

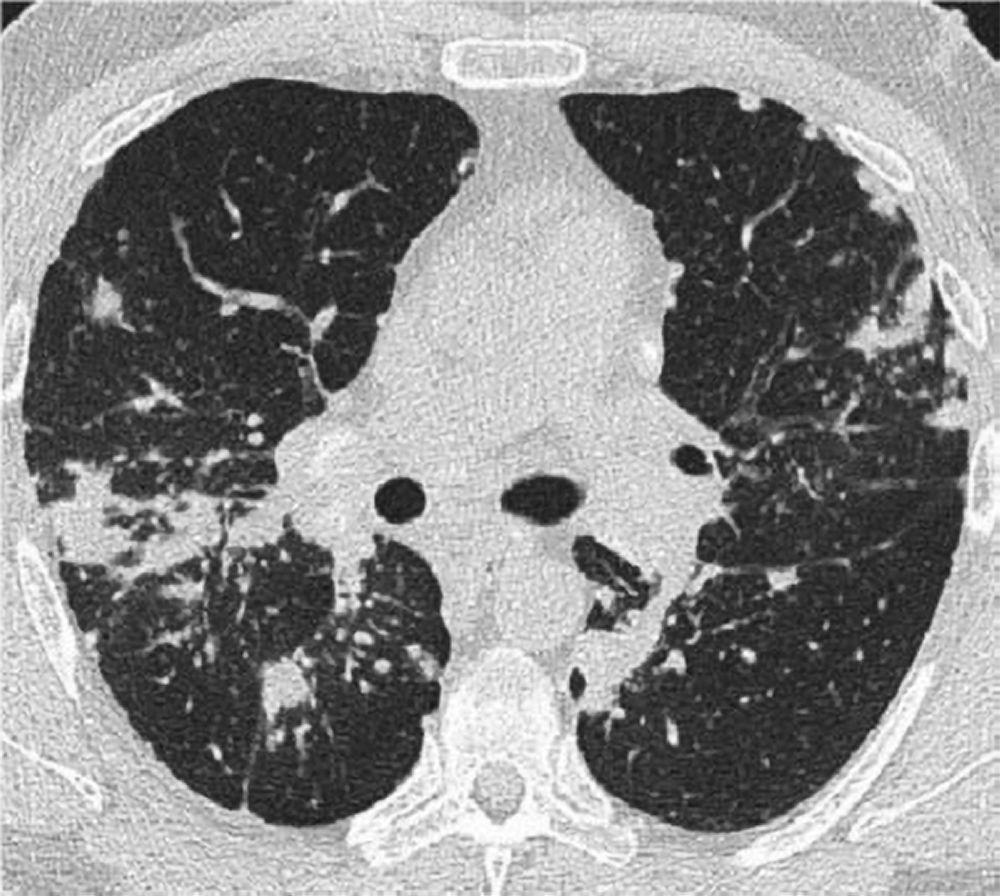
Image courtesy of Birendra P. Sah, MD, FCCP.
Quando a imagem sugere sarcoidose, confirmar o diagnóstico pela demonstração de granulomas sem caseificação na biópsia e exclusão de outras causas de doença granulomatosa (ver tabela Diagnóstico diferencial da sarcoidose). A síndrome de Löfgren não exige confirmação por biópsia.
A avaliação diagnóstica, portanto, requer:
Seleção do local da biópsia
Exclusão de outras causas de doença granulomatosa
Avaliação da gravidade e da extensão da doença para determinar se há indicação de tratamento
Locais para biópsia
Os locais para biópsia podem ser óbvios pelo exame físico e pela avaliação inicial: linfonodos periféricos, lesões dermatológicas e conjuntivas são facilmente acessíveis. Reportou-se que a punção aspirativa por agulha guiada por ultrassom endobrônquico (EBUS-TBNA, na sigla em inglês) de um linfonodo mediastinal ou hilar resulta em um diagnóstico de aproximadamente 90% e é o procedimento diagnóstico de escolha em pacientes com envolvimento intratorácico.
Quando há suspeita de sarcoidose pulmonar e o EBUS-TBNA não é diagnóstico, pode-se utilizar biópsia pulmonar transbrônquica broncoscópica com lavado broncoalveolar (LBA). Também pode ser utilizada em pacientes sem infiltrado parenquimatoso pulmonar porque o rendimento diagnóstico da biópsia pulmonar transbrônquica na sarcoidose em estádio I é de cerca de 50%. Se a biópsia transbrônquica broncoscópica não for diagnóstica, ela pode ser tentada uma segunda vez.
Se a EBUS-TBNA e biópsias transbrônquicas broncoscópicas não forem diagnósticas ou se a broncoscopia não puder ser tolerada, pode-se fazer uma mediastinoscopia para realizar uma biópsia dos linfonodos hilares ou mediastinais, ou também pode ser feita uma biópsia pulmonar toracoscópica assistida por vídeo (VAT) ou uma biópsia pulmonar aberta para coletar tecido pulmonar. Se há forte suspeita de sarcoidose, mas um local para realizar a biópsia não é evidente com base nos achados de exame ou imagem, a tomografia por emissão de pósitrons (PET) pode ajudar a identificar os locais ativos ocultos como coração, osso, músculo e encéfalo.
Exclusão de outros diagnósticos
A exclusão de outros diagnósticos é crucial, especialmente quando os sinais e sintomas da radiografia são mínimos, porque muitas outras doenças e processos podem causar inflamação granulomatosa (ver tabela Diagnóstico diferencial da sarcoidose). O tecido de biópsia deve ser submetido à cultura para fungos e micobactérias. Deve-se obter a história de exposição a antígenos ocupacionais (p. ex., silica, berílio), ambientais (p. ex., feno mofado, pássaros e outros deflagradores antigênicos de pneumonite por hipersensibilidade) e infecções (p. ex., tuberculose, coccidioidomicose, histoplasmose). Logo no início da avaliação, deve-se realizar o teste tuberculínico com derivado de proteína purificada (PPD) ou ensaio de liberação de interferon-gama.
Avaliação da gravidade da doença
Avalia-se a gravidade de acordo com o envolvimento de órgãos, por exemplo, somente com envolvimento pulmonar
Testes de função pulmonar
Os resultados dos testes de função pulmonar são frequentemente normais nos estágios iniciais, mas demonstram restrição e baixa difusão do monóxido de carbono (DLCO) na doença avançada. Também ocorre obstrução do fluxo aéreo, podendo sugerir o comprometimento da mucosa brônquica. Adicionar um teste de caminhada de 6 minutos pode caracterizar o comprometimento funcional de forma mais abrangente do que os resultados dos testes de função pulmonar isolados. Pacientes com comprometimento pulmonar extenso podem apresentar saturação normal de oxigênio em repouso, mas podem mostrar dessaturação aos esforços.
Para a detecção rotineira de doença extrapulmonar, recomendam-se os seguintes exames de triagem
ECG de 12 derivações, monitoramento com Holter, ecocardiografia
Exame oftalmológico com lâmpada de fenda
Exames de sangue de rotina para avaliar as funções renal e hepática
Níveis de cálcio sérico e excreção de cálcio na urina de 24 horas
Exames de imagem é muitas vezes necessária para detectar sarcoidose extrapulmonar. Ressonância magnética (RM) cardíaca com ou sem contraste com gadolínio pode ser apropriada para os pacientes com sintomas cardíacos. Em pacientes com sintomas neurológicos, pode ser necessária RM do encéfalo ou da medula espinal com ou sem contraste com gadolínio. Cintilografias ósseas e eletroneuromiografias podem ser apropriadas para pacientes com sintomas reumáticos. O PET scan parece ser o teste mais sensível para a detecção de sarcoidose óssea e outras sarcoidoses extrapulmonares e é utilizado em conjunto com a RM em pacientes com envolvimento cardíaco. Não se recomenda rotineiramente TC com agente de contraste radiopaco, mas pode fornecer evidências de comprometimento hepático ou esplênico (p. ex., aumento de volume e lesões hipolucentes). A cintilografia de corpo inteiro com gálio substituiu em grande medida a PET. Se a cintilografia com gálio de corpo inteiro está disponível, ela pode fornecer evidências fundamentais e úteis na ausência de confirmação tecidual. O aumento simétrico da captação dos linfonodos hilares e mediastinais (sinal de lambda) e das glândulas lacrimais, parótidas e salivares (sinal de panda) sugerem fortemente sarcoidose. O resultado negativo em pacientes que tomam prednisona não é confiável.
Os exames laboratoriais desempenham um papel adjuvante na determinação do diagnóstico e da extensão do envolvimento de órgãos. Hemograma completo com contagem diferencial pode evidenciar anemia, eosinofilia ou leucopenia. Deve-se medir o cálcio sérico para detectar hipercalcemia. Nitrogênio da ureia sanguínea, creatinina e exames de função hepática podem estar elevados nas sarcoidoses renal e hepática. A proteína total pode estar elevada em decorrência de hipergamaglobulinemia. A elevação da taxa de sedimentação de eritrócitos (velocidade de hemossedimentação) é comum, mas inespecífica. Recomenda-se a avaliação do cálcio em amostra urinária coletado ao longo de 24 horas para excluir hipercalciúria, mesmo em pacientes com níveis normais de cálcio sérico. A elevação dos níveis séricos de enzima conversora da angiotensina (ECA) pode sugerir sarcoidose, mas é inespecífica e esses níveis podem ser altos em pacientes com outras doenças (p. ex., hipertireoidismo, diabetes, doença de Gaucher, silicose, doença micobacteriana, infecções fúngicas, pneumonite por hipersensibilidade, linfoma). Se elevados, os níveis de enzima conversora de angiotensina (ECA) podem ser úteis para monitorar a adesão ao tratamento com corticoides. Os níveis da ECA diminuiem muito mesmo quando os pacientes tomam baixas doses de corticoides.
Deve-se incluir uma lavado broncoalveolar na biópsia broncoscópica para descartar infecções suspeitas (p. ex., quando achados de métodos menos invasivos, como exames de imagem, não são típicos da sarcoidose) e para excluir outros tipos de doença pulmonar intersticial se o diagnóstico de sarcoidose for duvidoso. Achados no lavado broncoalveolar variam consideravelmente, mas linfocitose (linfócitos > 15%) e/ou uma razão de CD4 +/CD8+> 3,5 na contagem diferencial das células do líquido do lavado sugerem o diagnóstico na vigência de contexto clínico apropriado. Entretanto, a ausência desses achados clínicos não exclui a sarcoidose.
Tratamento da sarcoidose
Anti-inflamatórios não esteroides (AINEs)
Corticoides
Imunossupressores
Anticorpos antifator de necrose tumoral alfa
Como a sarcoidose com frequência regride espontaneamente, os pacientes assintomáticos e aqueles com sintomas leves não necessitam de tratamento, embora devam ser monitorados para a identificação de sinais de deterioração. Esses pacientes podem ser seguidos com radiografias de tórax seriadas, testes de função pulmonar (incluindo a capacidade de difusão) e indicadores de envolvimento extratorácico (p. ex., exames rotineiros de função renal e hepática, exame oftalmológico anual com lâmpada de fenda). A frequência dos testes de acompanhamento é determinada pela gravidade da doença.
Pacientes que exigem tratamento independentemente do estágio das radiografias incluem aqueles com:
Piora dos sintomas
Limitação da atividade
Funções pulmonares deterioradas ou muito comprometidas
Alterações radiográficas preocupantes (p ex., avitação, fibrose, massas conglomeradas, sinais de hipertensão pulmonar)
Comprometimento do coração, do sistema nervoso, ou dos olhos
Insuficiência hepática ou renal
Hipercalcemia moderada a grave
Desfiguração da pele (p. ex., pus pernio) e doença articular
Os anti-inflamatórios não esteroides (AINEs) são utilizados para tratar o desconforto musculoesquelético.
Corticoides
Inicia-se o tratamento dos sintomas com corticoides. A presença de anormalidades nos exames torácicos de imagem sem sintomas significativos ou evidência de declínio na função do órgão não é uma indicação para o tratamento. Um protocolo padrão é prednisona, 20 mg a 40 mg/kg, por via oral uma vez ao dia, dependendo dos sintomas e da gravidade dos achados. Pode-se utilizar esquemas em dias alternados; p. ex., prednisona 40 mg por via oral 1 vez por dia, em dias alternados. Embora os pacientes raramente precisem de > 40 mg/dia, podem ser necessárias doses mais elevadas para reduzir as complicações na doença neurológica e cardíaca grave. A resposta geralmente ocorre dentro de 6 a 12 semanas, assim os sintomas, outros marcadores de gravidade da doença e os resultados dos testes de função pulmonar podem ser reavaliados entre 6 e 12 semanas. Casos insidiosos e crônicos podem responder mais lentamente. Corticoides são progressivamente reduzidos até a dose de manutenção (p. ex., prednisona 10 a 15 mg/dia) após evidência de resposta, e são mantidos por 6 a 9 meses adicionais se houver melhora.
A duração ótima do tratamento é desconhecida. A redução prematura da dose pode resultar em recidiva. Diminui-se de modo gradativo a dose do fármaco se não houver resposta ou se esta for equívoca. Finalmente, podem-se interromper os corticoides na maioria dos pacientes, mas como pode haver recaída em até 50% das vezes, deve-se repetir o monitoramento geralmente a cada 3 a 6 meses. Deve-se retomar o tratamento com corticoide na recidiva dos sinais e sintomas. Como a produção de enzima conversora da angiotensina (ECA) é suprimida com corticoides em doses baixas, a mensuração em série dos níveis séricos de ECA pode ser útil para avaliar a adesão ao uso de corticoides em pacientes com níveis elevados de ECA.
Corticoides inalatórios podem aliviar a tosse em pacientes com envolvimento endobrônquico. Um broncodilatador inalável pode ser adicionado em pacientes com doença obstrutiva das vias respiratórias.
Os corticoides tópicos podem ser úteis em alguns casos de doenças dermatológicas e oculares e em seios nasais.
Recomenda-se profilaxia contra pneumonia por Pneumocystis jirovecii enquanto os pacientes estiverem em uso de > 20 mg de prednisona por dia ou seu equivalente por mais de um mês e para aqueles sob tratamento com imunossupressores.
O alendronato ou outro bisfosfonato pode ser o tratamento de escolha na prevenção da osteoporose induzida por corticoides em pacientes de risco (p. ex. idosos). O uso suplementar de cálcio ou vitamina D comporta risco de hipercalcemia pela produção endógena da vitamina D ativa (1, 25 di-hidroxi vitamina D) pelos granulomas sarcoides. As dosagens do cálcio sérico e na urina de 24 horas devem estar normais antes de iniciar esses suplementos.
Dicas e conselhos
|
Imunossupressores
Imunossupressores são utilizados quando
Os pacientes não toleram prednisona
A sarcoidose é refratária a doses moderadas a altas de prednisona
A dose de prednisona não pode ser reduzida abaixo de 10 a 15 mg por dia após 3 meses
Antes do acréscimo de outros imunossupressores, deve-se considerar as possíveis razões para a falta de melhora clínica, como não adesão, doença comórbida (p. ex., asma, insuficiência cardíaca, anemia), hipertensão pulmonar, e fibrose em estágio terminal.
O metotrexato é o imunossupressor mais comumente utilizado. Os pacientes devem passar por um ensaio de 6 meses do metotrexato de 10 a 15 mg/semana. Antes de iniciar o metotrexato, os pacientes devem ser testados para infecção por vírus da hepatite B e vírus da hepatite C. Inicialmente, administram-se tanto corticoides como metotrexato; ao longo de 6 a 8 semanas, pode-se diminuir gradativamente a dose de corticoide e, em muitos casos, interrompê-la. No entanto, a resposta máxima ao metotrexato pode demorar 6 a 12 meses. Nesses casos, a dose de prednisona pode ser diminuída de modo mais lento. De início, realizam-se hemogramas e exames de função hepática seriados a cada 2 a 4 semanas e, em seguida, a cada 6 a 12 semanas, uma vez alcançada a dose estável. Recomenda-se a prescrição de ácido fólico (1 mg por via oral uma vez ao dia) para pacientes tratados com metotrexato para reduzir o risco de efeitos adversos.
Outros imunossupressores incluem azatioprina, micofenolato, ciclofosfamida, leflunomida e hidroxicloroquina. Hidroxicloroquina, 400 mg por via oral uma vez ao dia, ou 200 mg por via oral duas vezes ao dia, pode ser efetiva para o tratamento de hipercalcemia, artralgia, sarcoidose da pele ou linfonodos periféricos aumentados, desfigurantes ou desconfortáveis. Deve-se fazer a avaliação oftálmica antes do início da hidroxicloroquina e a cada 6 a 12 meses durante o tratamento para monitorar sua toxicidade ocular.
A recidiva é comum após a interrupção de um imunossupressor.
Anticorpos antifator de necrose tumoral alfa
O infliximabe é geralmente utilizado como medicação de 3ª linha para tratar a sarcoidose refratária e para tratar pacientes que são intolerantes tanto aos corticoides como aos imunossupressores mencionados previamente. Antes de iniciar o tratamento, os pacientes devem fazer um teste com derivado de proteína purificada (PPD) ou um ensaio de liberação de interferon gama para rastrear tuberculose latente. Infliximab é administrado por via intravenosa 3 a 5 mg/kg uma vez, novamente 2 semanas mais tarde, então 1 vez por mês. Pode levar 3 a 6 meses para alcançar a resposta máxima. O infliximabe é geralmente combinado com metotrexato ou azatioprina em baixas doses para prevenir a formação de anticorpos contra ele.
Pode-se considerar o uso de adalimumabe para pacientes com sarcoidose ocular ou cutânea e em pacientes que foram tratados com sucesso com infliximabe, mas que desenvolveram anticorpos ou reações à infusão. Adalimumabe, 40 a 80 mg, por via subcutânea, é administrado a cada 1 a 2 semanas.
Outras considerações de tratamento
Pacientes com bloqueio cardíaco ou arritmias ventriculares devido a comprometimento cardíaco devem receber um desfibrilador cardíaco implantável e um marca-passo, bem como tratamento farmacológico.
Nenhum fármaco disponível foi capaz de prevenir consistentemente a fibrose pulmonar.
O tratamento da hipertensão arterial pulmonar associada à sarcoidose (HAPS) é de suporte com diurese e oxigênio suplementar. O papel dos vasodilatadores pulmonares no tratamento da HPHS não está bem estabelecido; alguns estudos pequenos sugeriram eficácia, mas são necessários estudos maiores para confirmá-la (1).
O transplante de órgão é uma opção para pacientes com envolvimento pulmonar, cardíaco ou hepático em último estágio, embora a doença possa recidivar no órgão transplantado.
Pacientes com sarcoidose e comprometimento moderado ou grave da função pulmonar ou cardíaca têm risco aumentado de desenvolver tipos graves de infecção por covid-19 e de mortalidade. Como em pacientes com outras doenças pulmonares inflamatórias, durante a pandemia de covid-19, deve-se utilizar imunossupressores criteriosamente. A vacinação contra SARS-CoV-2 é fortemente recomendada em pacientes com sarcoidose porque pode reduzir a mortalidade e a gravidade da doença decorrente da infecção por covid-19. Um ensaio clínico em andamento visa avaliar melhor a eficácia da vacinação contra SARS-CoV-2 em pacientes com sarcoidose e em indivíduos em uso de imunossupressores.
Referência sobre o tratamento
1. Humbert M, Kovacs G, Hoeper MM, et al: 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J 61(1): 1-144, 2023. doi:10.1183/13993003.00879-2022
Prognóstico da sarcoidose
Embora a remissão espontânea seja comum, a gravidade e as manifestações da doença são altamente variáveis e muitos pacientes precisam de tratamento com corticoides para aliviar os sintomas ou desacelerar o declínio progressivo da função do órgão ou órgãos afetados em algum momento no curso da doença. Dessa maneira, o monitoramento seriado para a verificação de evidências de recaída é imperativo. Quase dois terços dos pacientes com sarcoidose acabam alcançando a remissão com poucas ou nenhuma sequela. Cerca de 50% dos pacientes que desenvolvem remissão espontânea o fazem dentro dos 3 primeiros anos após o diagnóstico. Menos de 10% desses pacientes apresentam recidiva depois de 2 anos. Aqueles que não desenvolvem remissão dentro de 2 a 3 anos têm probabilidade de ter doença crônica.
Admite-se que a sarcoidose seja crônica em até 30% dos pacientes e 10 a 20% desenvolvem sequelas permanentes. A doença é fatal em 1 a 5% dos pacientes, tipicamente devido à insuficiência respiratória causada por fibrose pulmonar e, menos frequente, por causa de hemorragia pulmonar provocada por aspergiloma. Entretanto, no Japão, a cardiomiopatia infiltrativa acarreta insuficiência cardíaca e arritmias, constituindo a causa mais comum de óbito.
O prognóstico é pior para os pacientes com sarcoidose extrapulmonar e para os negros. A remissão ocorre em 89% dos brancos e em 76% dos negros sem doença extratorácica e em 70% dos brancos e 46% dos negros com doença extratorácica.
Os sinais de prognóstico favorável incluem:
Síndrome de Löfgren (tríade de poliartrite aguda, eritema nodoso e adenopatia hilar)
Os sinais de prognóstico reservado incluem:
Uveíte crônica
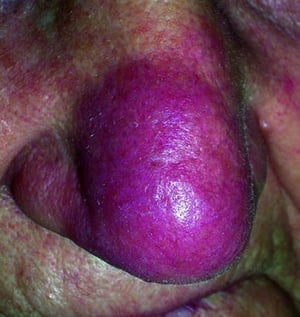
Lúpus pérnio
Hipercalcemia crônica
Neurossarcoidose
Envolvimento cardíaco
Envolvimento pulmonar extenso e/ou desenvolvimento de hipertensão pulmonar
Há pouca diferença na evolução a longo prazo entre os pacientes submetidos e os não submetidos ao tratamento e a recaída é comum ao término do tratamento.
Pontos-chave
O envolvimento sistêmico e extrapulmonar são comuns na sarcoidose, mas > 90% dos pacientes adultos têm comprometimento pulmonar.
Obter exames de imagem do tórax, mas confirmar o diagnóstico por biópsia, geralmente a aspiração transbrônquica guiada por ultrassom endobrônquico de um linfonodo mediastinal ou hilar.
Avaliar a gravidade do envolvimento pulmonar com testes da função pulmonar e oximetria de pulso durante exercícios.
Testar se há envolvimento extrapulmonar com ECG, exame com a lâmpada de fenda, testes de função renal e hepática, e testes de cálcio sérico e urinário.
Tratar pacientes com corticoides sistémicos quando indicado (p. ex., sintomas graves, hipercalcemia, declínio progressivo na função de órgãos, envolvimento cardíaco ou neurológico).
Tratar com imunossupressores se os pacientes não forem capazes tolerar doses moderadas de corticoides, se a sarcoidose for resistente a corticoides, ou se os corticoides forem necessários a longo prazo.







