




A glomerulonefrite rapidamente progressiva é uma síndrome nefrítica aguda acompanhada de formação de crescentes glomerulares microscópicos com progressão para falência renal em semanas a meses. O diagnóstico baseia-se em história, exame de urina, exames sorológicos e biópsia renal. O tratamento é realizado com corticoides, com ou sem ciclofosfamida ou rituximabe, e, às vezes, com troca plasmática.
O diagnóstico de glomerulonefrite rapidamente progressiva (GNRP), um tipo de síndrome nefrítica, é patológico e acompanhado por extensa formação de crescentes glomerulares (isto é, > 50% dos glomérulos amostrados contêm crescentes que podem ser observados na biópsia); se não tratada, a GNRP evolui para doença renal terminal dentro de semanas a meses. É relativamente incomum, afetando 10 a 15% dos pacientes com glomerulonefrite, e ocorre predominantemente entre 20 e 50 anos de idade. Classificam-se os tipos e as causas de acordo com os achados observados na microscopia imunofluorescente e testes serológicos (p. ex., anticorpos anti-MBG [GBM, anti-glomerular basement membrane], anticorpos citoplasmáticos antineutrófilos [ANCA] — ver tabela Classificação da glomerulonefrite rapidamente progressiva com base em microscopia por imunofluorescência).
Doença do anticorpo antimembrana basal antiglomerular
A doença do anticorpo antimembrana basal glomerular (MBG) é uma glomerulonefrite autoimune responsável por 10% dos casos de GNRP. Pode surgir quando a exposição respiratória (p. ex., fumaça de cigarros, infecções da via respiratória superior virais) ou a alguns outros estímulos expõe o colágeno dos capilares alveolares, desencadeando a formação de anticorpos anticolágeno. Os anticorpos anticolágeno apresentam reação cruzada com a MBG, fixando o complemento e desencadeando uma resposta inflamatória mediada por células nos rins e geralmente nos pulmões.
O termo síndrome de Goodpasture refere-se a uma combinação de glomerulonefrite e hemorragia alveolar na presença de anticorpos anti-MBG. Glomerulonefrite sem hemorragia alveolar na presença de anticorpos anti-MBG é chamada glomerulonefrite anti-MBG. A coloração por imunofluorescência da biópsia de tecido renal demonstra depósitos lineares de IgG.
Glomerulonefrite rapidamente progressiva por imunocomplexos
GNRP por imunocomplexos complica numerosas infecções e doenças reumáticas sistêmicas e também ocorre em outras glomerulopatias primárias.
A coloração na imunofluorescência demonstra depósitos granulares imunes inespecíficos. Essa condição contribui para 40% dos casos de glomerulonefrite rapidamente progressiva. A patogênese geralmente é desconhecida.
Glomerulonefrite rapidamente progressiva pauci-imune
A glomerulonefrite rapidamente progressiva pauci-imune é diferenciada pela ausência de imunocomplexos ou deposição de complemento na coloração por imunofluorescência. Constitui até 50% de todos os casos de glomerulonefrite rapidamente progressiva. Quase todos os pacientes apresentam anticorpos citoplasmáticos antineutrófilos elevados (ANCAs, geralmente ANCA-3 antiproteinase ou ANCA-mieloperoxidase) e vasculite sistêmica.
Doença de anticorpos duplos
A doença de anticorpos duplos ocorre com a presença de anticorpos anti-MBG e ANCA. É rara.
Glomerulonefrite rapidamente progressiva idiopática
Os casos idiopáticos são raros. Incluem pacientes com qualquer um dos seguintes:
Imunocomplexos, mas sem causa óbvia como infecção, doença reumática sistêmica, ou doença glomerular
Características pauci-imunes, mas ausência de anticorpos ANCA
Sinais e sintomas da glomerulonefrite rapidamente progressiva
A apresentação geralmente é insidiosa, com fraqueza, fadiga, febre, náuseas, vômitos, anorexia, artralgia e dor abdominal. Alguns pacientes apresentam semelhança com a glomerulonefrite pós-infecciosa com hematúria de início abrupto. Cerca de 50% dos pacientes apresentam edema e história de doença aguda semelhante à influenza cerca de 4 semanas antes do início da insuficiência renal, geralmente seguida de oligúria intensa. A síndrome nefrótica está presente em 10 a 30%. A hipertensão é pouco comum e raramente grave. Pacientes com doença de anticorpos anti-MBG podem ter hemorragia pulmonar, que pode se apresentar como hemoptise ou ser detectável apenas por infiltrados alveolares difusos nos exames de imagem pulmonares (síndrome pulmonar-renal ou síndrome de hemorragia alveolar difusa).
Diagnóstico da glomerulonefrite rapidamente progressiva
Insuficiência renal progressiva ao longo de semanas ou meses
Sedimento urinário nefrítico
Exames sorológicos
Níveis séricos do complemento
Biópsia renal
O diagnóstico é sugerido por lesão renal aguda em pacientes com hematúria e eritrócitos dismórficos ou cilindros hemáticos. Os exames incluem creatinina sérica, exame de urina, hemograma completo, testes sorológicos e biópsia renal. O diagnóstico habitualmente é feito pelos testes sorológicos e pela biópsia renal.
A creatinina sérica está quase sempre elevada.
O exame de urina mostra que hematúria sempre está presente, e cilindros hemáticos normalmente estão presentes. Sedimentos “telescópicos” (isto é, sedimento com múltiplos elementos, incluindo leucócitos, eritrócitos dismórficos e cilindros leucocitários, hemáticos, granulares, serosos e grandes) são comuns.
No hemograma anemia sempre está presente, e leucocitose é comum.
Os exames sorológicos devem incluir anticorpos anti- MBG (doença de anticorpos anti- MBG); anticorpos antiestreptolisina O, anticorpos anti- DNA ou crioglobulinas (glomerulonefrite rapidamente progressiva por complexos imunitários); e títulos de anticorpo citoplasmático antineutrófilo (ANCA) (glomerulonefrite rapidamente progressiva pauci-imune).
A dosagem do complemento (C3 e C4 sérico) pode ser útil se houver suspeita de glomerulonefrite rapidamente progressiva por imunocomplexos porque hipocomplementemia é comum.
A biópsia renal precoce é essencial. A característica comum a todos os tipos de glomerulonefrite rapidamente progressiva é a proliferação focal de células epiteliais glomerulares, algumas vezes entremeadas por numerosos neutrófilos, que formam uma massa celular em crescente (crescentes) que preenche o espaço de Bowman em > 50% dos glomérulos. O tufo glomerular geralmente aparece hipocelular e colapsado. Pode haver necrose no interior do tufo ou envolvendo o crescente, podendo ser a anormalidade mais proeminente. Nesses pacientes, devem-se pesquisar evidências histológicas de vasculite.

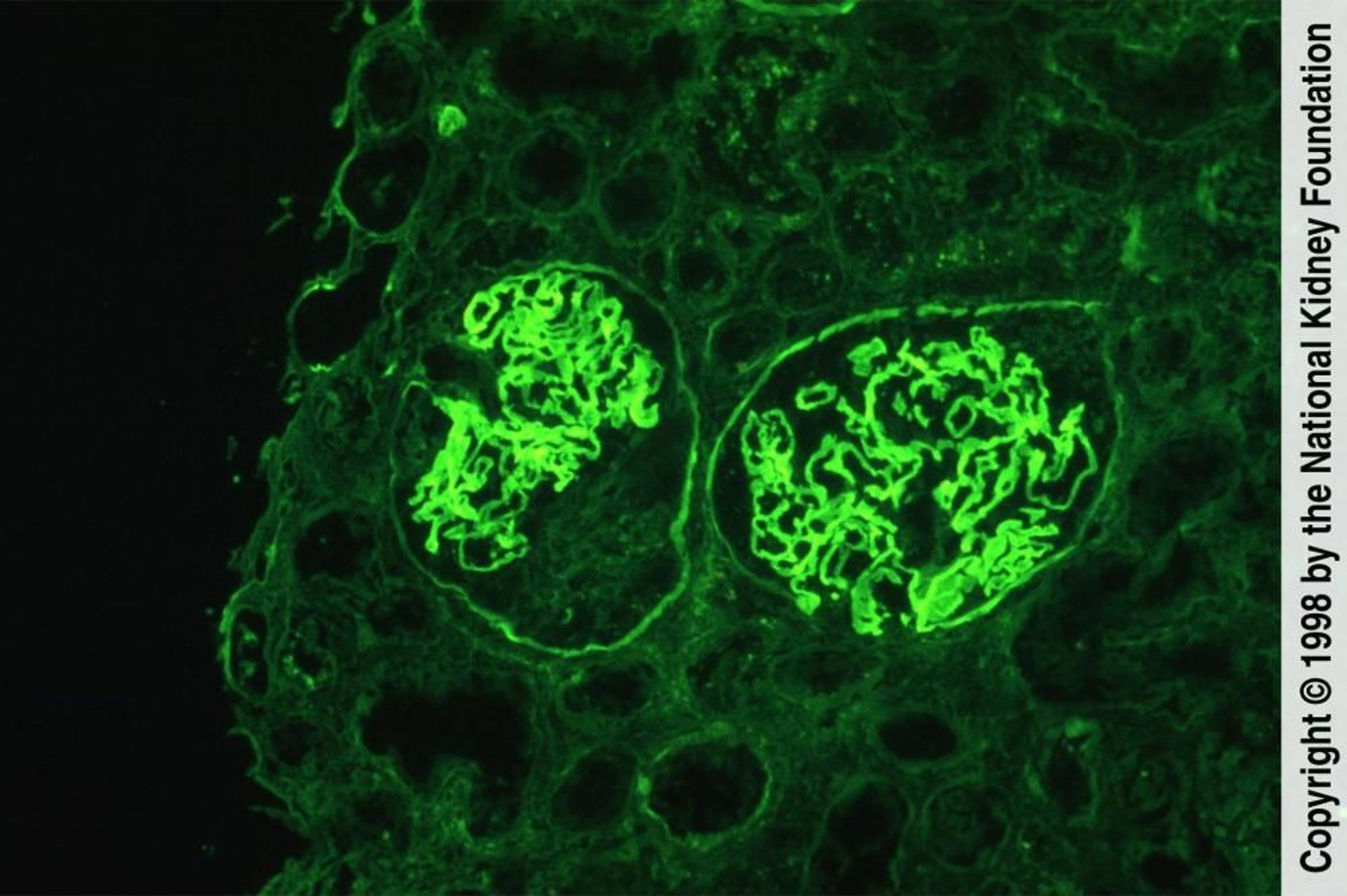
Imagem fornecida por Agnes Fogo, MD, e the American Journal of Kidney Diseases' Atlas of Renal Pathology (ver www.ajkd.org).
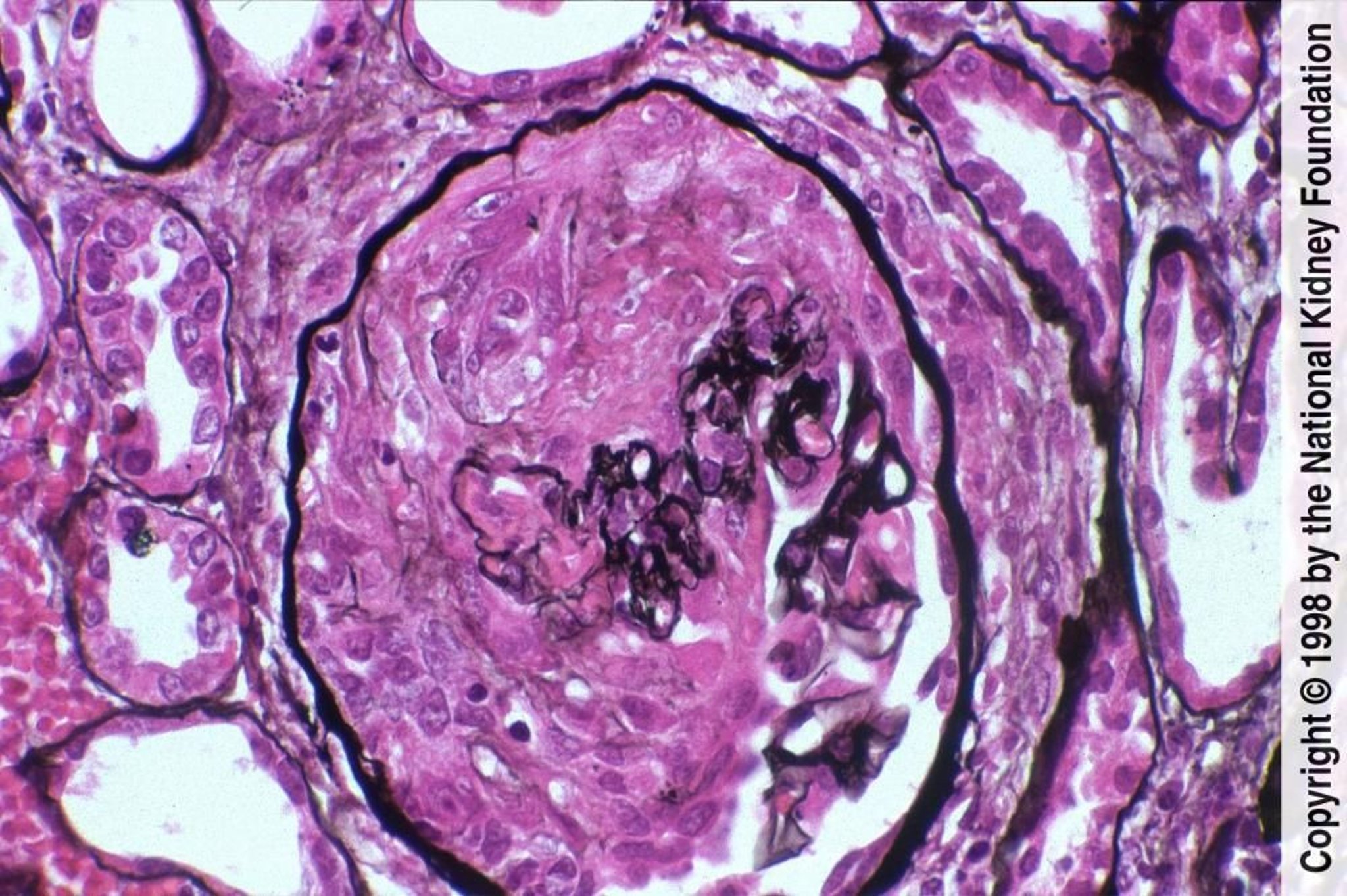
Image provided by Agnes Fogo, MD, and the American Journal of Kidney Diseases' Atlas of Renal Pathology (see www.ajkd.org).
Os resultados da microscopia com imunofluorescência diferem para cada tipo:
Na doença de anticorpos anti-MBG, a deposição linear ou em fita de IgG ao longo da MBG é mais proeminente e geralmente é acompanhada por deposição linear e algumas vezes granular de C3.
Na glomerulonefrite rapidamente progressiva por imunocomplexos, a imunofluorescência revela depósitos difusos mesangiais de IgG e C3, irregulares.
Na glomerulonefrite rapidamente progressiva pauci-imune, não são detectados coloração imunitária ou depósitos. Entretanto, há presença de fibrina nos crescentes, independentemente do padrão de fluorescência.
Na glomerulonefrite rapidamente progressiva de duplo anticorpo, há coloração linear da MBG.
Na glomerulonefrite rapidamente progressiva idiopática, alguns pacientes apresentam complexos imunitários e outros apresentam ausência de coloração imunitária e depósitos.
Tratamento da glomerulonefrite rapidamente progressiva
Corticoides
Ciclofosfamida
Rituximabe
Troca plasmática
O tratamento varia de acordo com o tipo da doença, apesar de não haver esquemas estudados com rigor. Deve ser instituído precocemente, de preferência quando a creatinina sérica estiver < 5 mg/dL (442 micromol/L) e antes da biópsia mostrar envolvimento em crescente de todos os glomérulos ou crescentes em organização, assim como também interstício fibrótico e atrofia tubular. Deve-se tratar mesmo os pacientes com comprometimento renal e níveis mais altos de creatinina agressivamente se não exigem tratamento de substituição renal. O tratamento torna-se menos eficaz na medida em que essas características se tornam mais proeminentes e pode ser lesivo em algumas populações (p. ex., pacientes idosos ou com infecção).
Corticoides e ciclofosfamida ou rituximabe costumam ser administrados. Para doença pauci-imune e imunocomplexo, corticoides (metilprednisolona, 1 g, intravenosa, uma vez ao dia, ao longo de 30 min, durante 3 a 5 dias, seguida de prednisona, 1 mg/kg, por via oral, uma vez ao dia) pode reduzir a creatinina sérica ou retardar a diálise por > 3 anos em 50% dos pacientes (1, 2).
Ciclofosfamida pode beneficiar pacientes positivos para ANCA (anticorpo citoplasmático antineutrófilo); os esquemas com pulsos mensais podem causar menos efeitos adversos (p. ex., leucopenia, infecção) do que a terapia oral devido à dosagem cumulativa reduzida. Normalmente introduz-se prednisona e ciclofosfamida concomitantemente à troca plasmática na doença de anticorpos anti- MBG (membrana basal glomerular), continuadas para minimizar a formação de novos anticorpos. Pacientes com doença idiopática são geralmente tratados com corticoides e ciclofosfamida, mas dispõem-se de poucos dados sobre a eficácia.
Rituximabe pode ser dosado em 375 mg/m2 por semana durante 4 semanas, como utilizado no estudo RAVE (título formal: Rituximab in ANCA-Associated Vasculitis; [2]). Um esquema alternativo é uma dose inicial de 1 g, seguida de outra dose de 1 g depois de 2 semanas. O rituximabe não tem sido utilizado no tratamento da doença anti- MBG.
Troca plasmática (trocas diárias de 3 a 4 L durante 14 dias) é recomendada para a doença de anticorpos anti-MBG. A troca plasmática também pode ser considerado ser considerada para glomerulonefrite rapidamente progressiva por imunocomplexos e pauci-imune associada a ANCA com hemorragia pulmonar ou disfunção renal grave na apresentação [creatinina sérica > 5 a 7 mg/dL (442 a 618,8 micromol/L) ou dependência de diálise], mas seu uso permanece controverso. Acredita-se que a troca plasmática remova rapidamente os anticorpos livres, os imunocomplexos intactos e os mediadores da inflamação (p. ex., fibrinogênio, complemento). Embora algumas evidências sugiram que a troca plasmática melhorou os resultados renais a curto prazo, um estudo randomizado subsequente não mostrou que reduziu a incidência de morte ou doença renal terminal (3).
Terapia imunossupressora agressiva também pode ser benéfica para pacientes com níveis mais elevados de creatinina. Plasmaferese combinada com prednisona e ciclofosfamida beneficiou pacientes com envolvimento renal que não exigem terapia de substituição renal imediata, mesmo que os níveis de creatinina estivessem acima de 5 a 7 mg/dL (442 a 618,8 micromol/L; [4]).
O transplante de rim é eficaz para todos os tipos, mas a doença pode reincidir no rim transplantado; o risco diminui com o tempo. Na doença por anticorpos anti-MBG, os títulos de anticorpos anti-MBG devem permanecer indetectáveis por pelo menos 12 meses antes do transplante. Para pacientes com glomerulonefrite rapidamente progressiva pauci-imune, a atividade da doença deve ser quiescente por pelo menos 6 meses antes do transplante; os títulos de ANCA não precisam ser suprimidos.
Referências sobre o tratamento
1. Ponticelli C, Altieri P, et al: A randomized study comparing methylprednisolone plus chlorambucil versus methylprednisolone plus cyclophosphamide in idiopathic membranous nephropathy. J Am Soc Nephrol 9(3):444, 1998. doi: 10.1681/ASN.V93444
2. Jones RB, Cohen Tervaert JW, Hauser T: Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis. N Engl J Med 363:211-220, 2010. doi: 10.1056/NEJMoa0909169
3. Walsh M, Merkel PA, Peh C-A, et al: Plasma exchange and glucocorticoids in severe ANCA-associated vasculitis. N Engl J Med 382(7):621-631, 2020. doi: 10.1056/NEJMoa1803537
4. Levy JB, Turner AN, Rees AJ, et al: Long-term outcome of anti-glomerular basement membrane antibody disease treated with plasma exchange and immunosuppression. Ann Intern Med 134(11):1033-1042, 2001. doi: 10.7326/0003-4819-134-11-200106050-00009
Prognóstico da glomerulonefrite rapidamente progressiva
A remissão espontânea é rara e 80 a 90% dos pacientes não tratados evoluem para doença renal em estágio terminal em 6 meses. O prognóstico melhora com o tratamento precoce.
Os fatores prognósticos favoráveis de resposta incluem glomerulonefrite rapidamente progressiva causada pelos seguintes:
Doença anti-MBG, se tratada precocemente, em especial quando tratada antes do surgimento de oligúria e nível de creatinina < 7 mg/dL (618,8 micromol/L)
Os fatores prognósticos desfavoráveis incluem os seguintes:
Idade > 60 anos
Insuficiência renal oligúrica
Nível mais alto de creatinina
Crescentes circunferenciais em > 75% dos glomérulos
Glomerulonefrite rapidamente progressiva pauci-imune
Cerca de 30% dos pacientes com glomerulonefrite rapidamente progressiva pauci-imune não respondem ao tratamento; entre aqueles que não respondem, 40% exigem diálise e cerca de 33% morrem em 4 anos. Em contraste, entre os pacientes que respondem ao tratamento, < 20% necessitam de diálise e ao redor de 3% morrem.
Pacientes com doença de anticorpos duplos parecem ter um prognóstico renal relativamente melhor do que pacientes apenas com a doença de anticorpos antimembrana basal e pior do que pacientes com doença pauci-imune.
Os pacientes que recuperam a função renal normal após glomerulonefrite rapidamente progressiva demonstram alterações histológicas residuais principalmente nos glomérulos, consistindo principalmente em hipercelularidade, com pouca ou nenhuma esclerose dentro do tufo glomerular ou das células epiteliais e fibrose mínima do interstício.
A morte habitualmente decorre de infecções ou causas cardíacas, desde que a morte por uremia seja evitada pela diálise.
Pontos-chave
Considerar glomerulonefrite rapidamente progressiva se os pacientes têm lesão renal aguda com hematúria e eritrócitos dismórficos ou cilindros hemáticos, particularmente na presença de sintomas constitucionais ou inespecíficos subagudos (p. ex., fadiga, febre, anorexia, artralgia, dor abdominal).
Avaliar com testes sorológicos e biópsia renal precoce.
Iniciar o tratamento precocemente com corticoides e ciclofosfamida e, em alguns casos, troca plasmática.
Considerar transplante renal após a atividade da doença ser controlada.







