




A poliangiite microscópica é uma vasculite necrosante sistêmica sem deposição de imunoglobulina (pauci-imune) que afeta principalmente os pequenos vasos. Ela pode começar como uma síndrome renal-pulmonar e rapidamente progredir para glomerulonefrite e hemorragia alveolar, mas o padrão da doença depende dos órgãos afetados. O diagnóstico é feito por resultados clínicos e às vezes confirmado por meio de biópsia. O tratamento depende da gravidade da doença, sendo feito com corticoides e imunossupressores.
(Ver também Visão geral da vasculite.)
Poliangiite microscópica (MPA) é rara (cerca de 13 a 19 casos/milhão). A patogênese é desconhecida. A poliarterite microscópica atinge os pequenos vasos, é pauci-imune (isto é, não se observa deposição de imunoglobulina no tecido da biópsia), semelhante à granulomatose com poliangiite e à granulomatose eosinofílica com poliangiite, o que a diferencia das vasculites de pequenos vasos mediadas por imunocomplexos (p. ex., vasculite associada à imunoglobulina A— anteriormente conhecida como púrpura de Henoch-Schönlein) e da vasculite cutânea de pequenos vasos. A MPA afeta predominantemente os pequenos vasos (incluindo capilares e vênulas pós-capilares), ao contrário da poliarterite nodosa, que afeta as artérias musculares de tamanho médio. A literatura mais antiga (isto é, antes de 1994) não diferenciava adequadamente a poliarterite nodosa da MPA — a hemorragia alveolar e a glomerulonefrite podem ocorrer na MPA, mas não na poliarterite nodosa. Raramente, a MPA pode ocorrer em associação à hepatite B.
As manifestações clínicas se parecem com aquelas da granulomatose com poliangiite, exceto pelo fato de as lesões granulomatosas e destrutivas (lesões cavitárias pulmonares) estarem ausentes e o trato respiratório superior geralmente ser afetado minimamente ou não ser absolutamente afetado. Nos dois distúrbios os anticorpos citoplasmáticos antineutrófilos (ANCA) podem estar presentes.
Sinais e sintomas da poliangeíte microscópica
Em geral, a fase prodrômica da doença com sintomas sistêmicos de febre, perda ponderal, mialgia e artralgia ocorre. Outros sintomas dependem de quais órgãos e sistemas estão afetados:
Renal: os rins são afetados em até 90% dos pacientes. Estão presentes hematúria, proteinúria, (às vezes > 3 g/24 horas) e cilindros hemáticos. Sem o exato diagnóstico e tratamento, a insuficiência renal pode progredir rapidamente.
Cutâneo: cerca de um terço dos pacientes tem um exantema purpúrico quando o diagnóstico é feito. Infartos no leito ungueal podem causar hemorragia subungueal; a isquemia digital raramente ocorre.
Respiratório: se os pulmões forem afetados, pode ocorrer hemorragia alveolar que pode ser seguida de fibrose pulmonar. Rápido início de dispneia e anemia, com ou sem hemoptise e infiltrados bilaterais irregulares (vistos na radiografia do tórax), podem ser resultado de hemorragia alveolar, uma emergência médica que requer tratamento imediato. Alguns pacientes com poliarterite microscópica podem inicialmente apresentar doença pulmonar intersticial. Sintomas leves de rinite, epistaxe e sinusite podem ocorrer; entretanto, se o trato respiratório superior estiver gravemente afetado, é mais provável que a causa seja granulomatose com poliangiite.
Gastrointestinal: os sintomas gastrointestinais são dor abdominal, náuseas, vômitos, diarreia e sangue nas fezes.
Neurológico: se o sistema nervoso for afetado, a mononeuropatia múltipla (mononeurite multipla) que afeta os nervos periféricos e cranianos normalmente ocorre. Raramente, hemorragia cerebral, infarto, convulsão ou cefaleia resultam de vasculite cerebral.
Cardíaco: O coração é raramente afetado.
Ocular: se os olhos forem afetados, geralmente ocorre episclerite.
Diagnóstico da poliangeíte microscópica
Achados clínicos
Testes para ANCA (antineutrophil cytoplasmic autoantibodies) e exames laboratoriais de rotina
Biópsia
Deve-se suspeitar de poliangiite microscópica em pacientes que apresentam combinações inexplicáveis de febre, perda ponderal, artralgias, dor abdominal, hemorragia alveolar, síndrome nefrítica de início recente, mononeuropatia de início recente ou polineuropatia. São realizados testes de laboratório e às vezes radiografias, mas geralmente o diagnóstico é confirmado por biópsia.
Os testes incluem hemograma, velocidade de hemossedimentação (velocidade de hemossedimentação), proteína C-reativa, exame de urina, creatinina sérica e teste para ANCA (antineutrophil cytoplasmic antibodies). Níveis de VHS, proteína C-reativa e contagem de leucócitos e plaquetas são elevados, refletindo inflamação sistêmica. Anemia de doença crônica é comum. Uma queda aguda no hematócrito sugere hemorragia alveolar ou hemorragia no trato gastrointestinal. A análise de urina (para verificar hematúria, proteinúria e cilindros celulares) deve ser feita e a creatinina sérica deve ser medida periodicamente para verificar envolvimento renal.
Teste de imunofluorescência pode detectar ANCA; este teste é seguido por testes de imunofluorescência e ensaio imunoadsorvente ligado à enzima (ELISA) para verificar anticorpos específicos. Pelo menos 60% dos pacientes são positivos para ANCA, geralmente ANCA perinuclear (p-ANCA) com anticorpos contra mieloperoxidase.
A biópsia do tecido envolvido mais acessível deve ser feita para confirmar vasculite. A biópsia renal pode detectar glomerulonefrite necrosante pauci-imune focal e segmentar, com necrose fibrinoide da parede do capilar glomerular levando a formação celular crescente.
Em pacientes com sintomas respiratórios, realiza-se exame por imagem do tórax para verificar infiltrados. Infiltrados bilaterais irregulares sugerem hemorragia alveolar mesmo em pacientes sem hemoptise. A TC é bem mais sensível do que a radiografia.

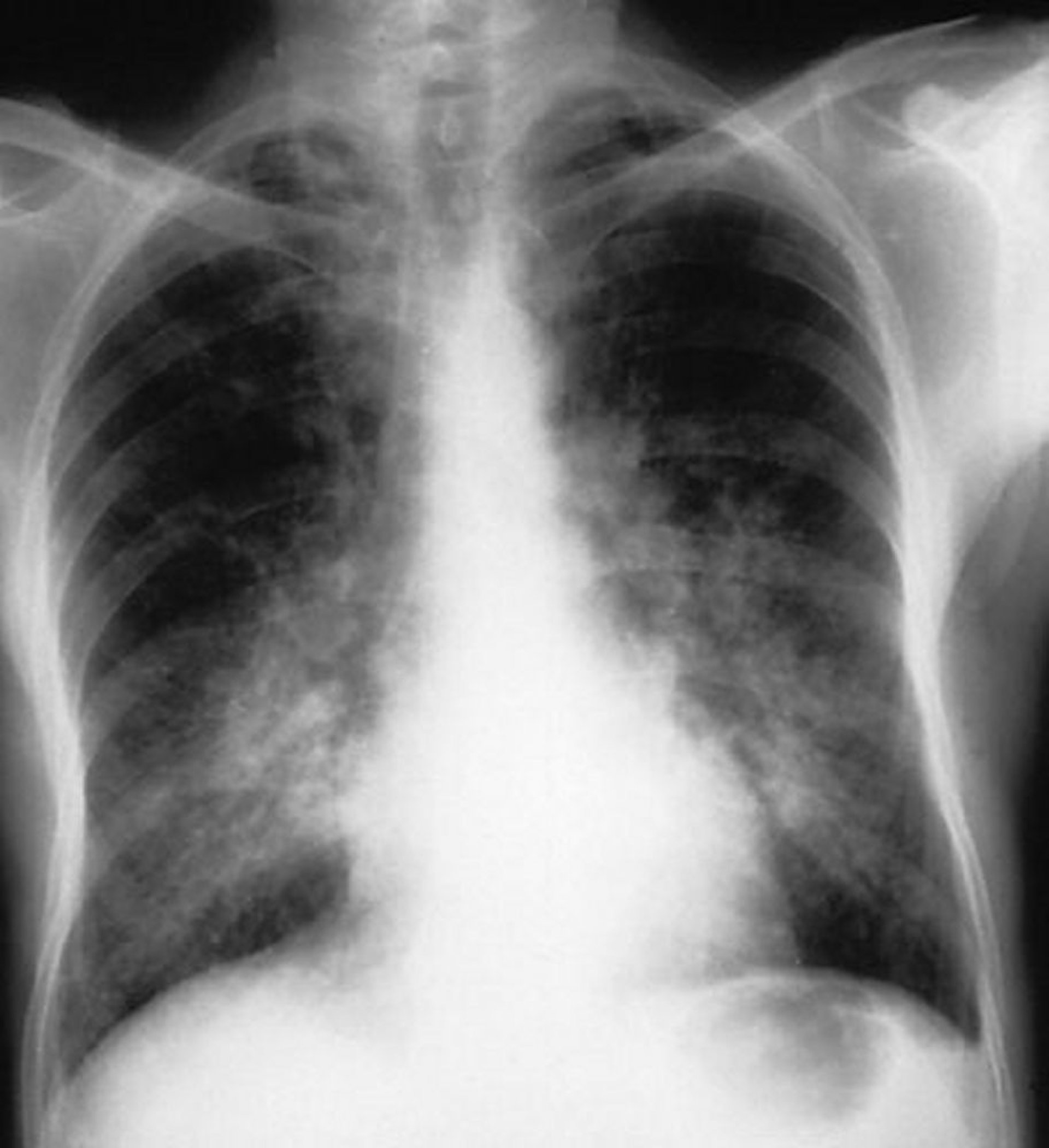
Com a permissão do editor. From Cohen A, Glassock R. In Atlas of Diseases of the Kidney: Glomerulonephritis and Vasculitis. Editado por R Schrier (editor da série), RJ Glassock, and AH Cohen. Philadelphia, Current Medicine, 1999.
Se os pacientes têm dispneia e infiltrados bilaterais, a broncoscopia deve ser feita imediatamente para verificar se há hemorragia alveolar e para excluir infecção. Sangue vindo de ambos os pulmões e todos os brônquios, com mais sangue vindo conforme o broncoscópio vai penetrando nas vias respiratórias, indica hemorragia alveolar ativa. Macrófagos corados com hemossiderina aparecem de 24 a 72 horas após o início da hemorragia e pode persistir por até 2 meses.
Tratamento da poliangeíte microscópica
Quando órgãos vitais são afetados, deve-se utilizar alto dose de corticoides mais ciclofosfamida ou rituximabe
Nos casos menos graves, corticoides e azatioprina ou metotrexato
O tratamento de indução da remissão é semelhante ao do granulomatose com poliangiite, mas a necessidade de tratamento de manutenção não está tão claramente estabelecida no poliangiite microscópica. A ciclofosfamida dada diariamente mais corticoides melhora a sobrevida quando órgãos vitais são afetados. Mostrou-se que o rituximabe não é inferior à ciclofosfamida para induzir a remissão da doença grave. Regimes de indução e manutenção variam e terapias adjuvantes, como troca plasmática e metilprednisolona pulsada IV podem ou não ser utilizadas.
Casos menos graves podem ser tratados com corticoides junto com metotrexato.
Pontos-chave
Poliangiite microscópica é uma vasculite rara dos pequenos vasos.
As manifestações são variáveis e podem incluir hemorragia alveolar, mononeuropatia múltipla e glomerulonefrite.
Confirmar o diagnóstico fazendo sorologia de ANCA (antineutrophil cytoplasmic antibodies) e biópsia.
Tratar com corticoides e um imunossupressor (p. ex., ciclofosfamida ou rituximabe para doença grave).







