




ハンチントン病は遺伝性疾患で、初期には不随意な筋肉のひきつりまたはけいれんがときおり起こり、進行すると顕著な不随意運動(舞踏運動とアテトーゼ)と精神機能の低下が現れ、死に至ります。
ハンチントン病では、動作を滑らかにして協調させる働きのある脳領域に変性が生じます。
動作がぎこちなくなって協調運動が難しくなり、自制や記憶などの精神機能が低下します。
診断は症状および家族歴と脳の画像検査および遺伝子検査の結果に基づいて下されます。
薬剤によって症状を軽減できますが、病気は進行性であり、最終的には死に至ります。
(運動障害の概要も参照のこと。)
ハンチントン病は10万人に1~10人の割合で発生します。患者の数は、世界の地域によって異なります。男女差はありません。
ハンチントン病の遺伝子は優性遺伝します。つまり、両親のどちらかから異常な遺伝子を1つ受け継ぐだけで、この病気を発症するということです。そのため、ハンチントン病がある人の子どもは50%の確率で発症します。
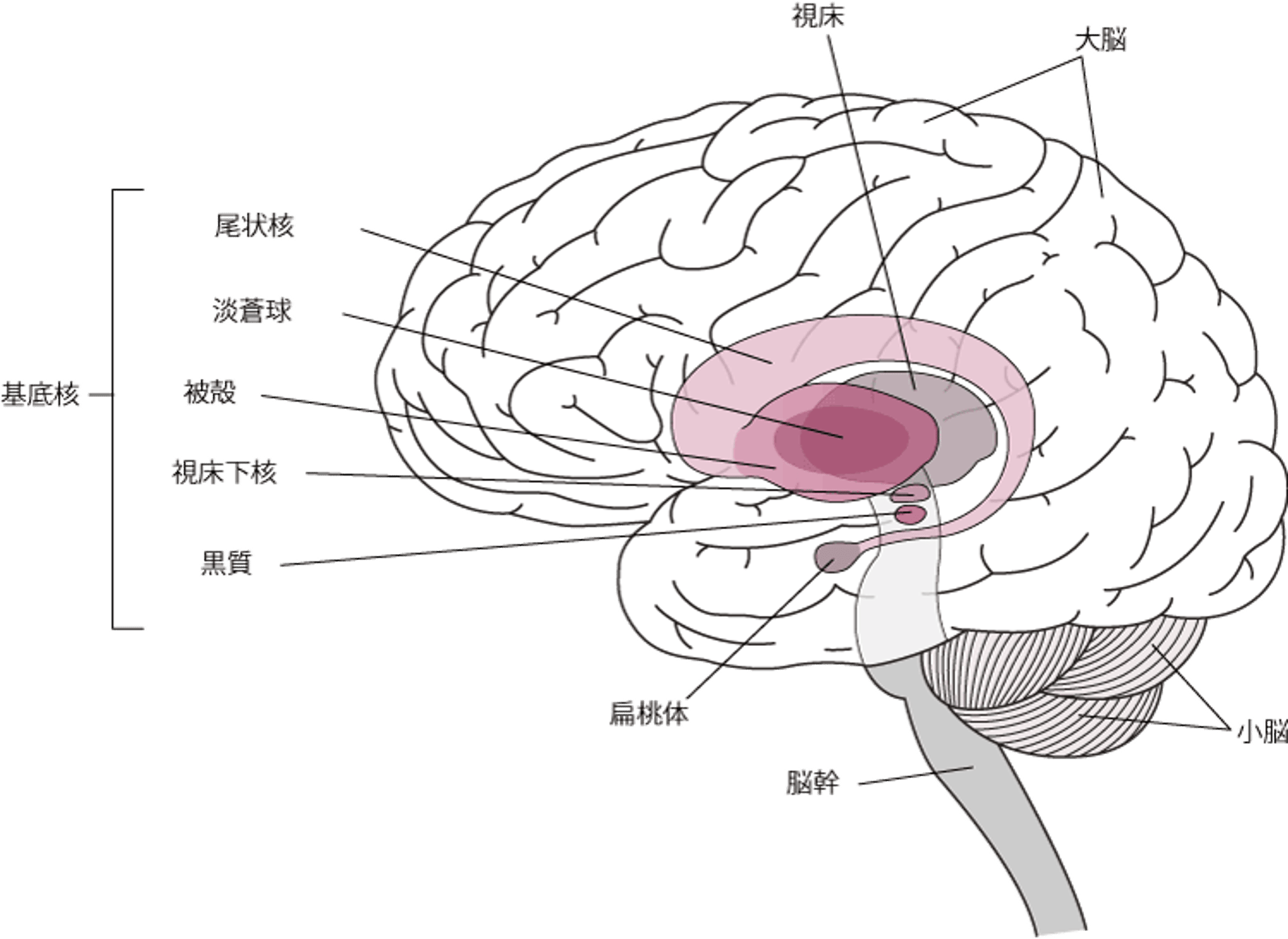
ハンチントン病の原因は、尾状核と被殻と呼ばれる大脳基底核の一部で徐々に起こる変性です。大脳基底核は、脳の奥深く、大脳の基底部にある神経細胞の集まりです。大脳基底核には、筋肉の動きを滑らかにして調整する働きがあります。
大脳基底核の位置
大脳基底核は、脳の奥深くにある神経細胞の集まりです。以下のものが含まれます。
大脳基底核には、筋肉の運動を開始し、その動きを滑らかにし、不随意運動を抑制し、姿勢の変化を調整する機能があります。 |

ハンチントン病の症状
ハンチントン病は通常、かすかな症状で始まります。発症年齢は一般に35~40歳ですが、成人前に発症することもあります。
ハンチントン病の初期には、顔面、体幹、四肢が意図せず急速に動くことがあります。初めは、こうした異常な不随意運動を意図的な動作の中に組み込むことができるため、異常な動きはほとんど気づかれません。しかし、時間が経つにつれ、動きが顕著になります。
筋肉が短く急速に収縮し、腕や別の部位が突然ビクッと(ときに何回か続けて)動くことがあります。
操り人形のように、軽く弾むような、あるいは過剰にはつらつとした歩行がみられます。しかめ面をする、腕や脚が弾むように動く、まばたきが頻繁になるなどの症状も現れます。協調運動が難しくなり、動作が遅くなります。最終的には全身に影響が及び、歩く、静かに座っている、食べる、話す、飲み込む、服を着るなどの動作が極めて困難になります。
多くの場合、異常な動きの発生前または発生と同時期に精神的な変化が生じますが、最初は目立ちません。徐々にいらだち、興奮、動揺が生じやすくなります。普段行っていた活動への興味が失われることもあります。衝動を抑えられない、怒りっぽい、発作的に落胆する、分別がなくなるなどの症状もみられます。
ハンチントン病が進行すると行動が無責任になり、しばしばあてもなく徘徊するようになります。数年経過すると、記憶が障害され合理的な思考ができなくなります。重度の抑うつ状態になり、自殺を試みることもあります。不安になったり、強迫症を発症したりすることもあります。
病気が進行すると、重度の認知症が生じ、寝たきりになります。24時間の介助か介護施設への入所が必要になります。多くの人は発症してから13~15年後に亡くなります。
ハンチントン病の診断
医師による評価と遺伝子検査による確定
CTまたはMRI検査
初期のハンチントン病は、症状がわずかで気づかれにくいことがあります。ハンチントン病は、症状と家族歴に基づいて疑われることがあります。精神的な問題がある人や、神経疾患(パーキンソン病など)または精神障害(統合失調症など)と診断された人が近親者にいる場合は、医師に伝えるべきです。これは、ハンチントン病でありながら、ハンチントン病と診断されていない可能性があるからです。
医師はCTまたはMRI検査を行って、大脳基底核やこの病気でよく侵される脳の領域に変性がないかを調べるとともに、他の病気の可能性を否定します。
診断を確定するために遺伝子検査が行われます。症状が現れる前に子どもができる可能性も高いため、ハンチントン病の家族歴があっても症状がない人にとって、遺伝子検査とカウンセリングを受けることは重要です。そのような人は、遺伝子検査を受ける前に遺伝カウンセリングを受けるべきです。その場合、複雑な倫理的・心理的問題に対処できる専門施設への紹介が行われます。
ハンチントン病の治療
抗精神病薬やその他の薬剤による症状の軽減
ハンチントン病と診断されたら、できるだけ早いうちに、終末期にどのような治療を望むかを示した事前指示書を作成しておくべきです。
ハンチントン病には根治的な治療法がありません。しかし、抗精神病薬(クロルプロマジン、ハロペリドール、リスペリドン、オランザピンなど)など、一部の薬剤は興奮の制御に役立つことがあります。ドパミンの量を減らす薬(テトラベナジン、デューテトラベナジン[deutetrabenazine]、降圧薬のレセルピンなど)は、異常な動きを止める(抑制する)のに役立ちます。
うつ病があれば、その治療のために抗うつ薬を使用できます。
医師はハンチントン病の親や兄弟姉妹がいる人に対して、遺伝カウンセリングと遺伝子検査を勧めます。ハンチントン病は経過がとても深刻な病気ですので、遺伝子検査の前に遺伝カウンセリングが勧められるべきです。妊娠可能年齢の女性と子どもをもつことを考えている男性には、カウンセリングが特に重要になります。
さらなる情報
役立つ可能性がある英語の資料を以下に示します。こちらの情報源の内容について、MSDマニュアルでは責任を負いませんのでご了承ください。
ジェネティクス・ホーム・リファレンス:ハンチントン病(Genetics Home Reference: Huntington disease):このウェブサイトには、ハンチントン病の説明や、その原因や遺伝形式に関する考察のほか、診断と治療に関するリンクも掲載されています。




