




アミロイドーシスは,異常凝集したタンパク質から成る不溶性線維の細胞外蓄積を特徴とする多様な疾患群である。これらのタンパク質は局所に蓄積してほとんど症状を引き起こさない場合もあるが,全身の複数の臓器に蓄積して,重度の多臓器不全をもたらすこともある。アミロイドーシスは原発性の場合と,種々の感染症,炎症,または悪性疾患に続発する場合とがある。診断は罹患組織の生検によるが,アミロイド原性タンパク質の病型の判別には種々の免疫組織学的および生化学的手法が用いられる。治療はアミロイドーシスの病型によって異なる。
アミロイド沈着物は以下のもので構成されている:
X線回折により確認可能な,βシート構造を形成する微小(直径約10nm)な不溶性線維
この沈着物には,線維状のアミロイドタンパク質に加えて血清アミロイドP成分およびグリコサミノグリカンも含まれる。
アミロイド線維は,凝集してオリゴマーを経て不溶性線維となったミスフォールディングタンパク質で構成される。こうしたミスフォールディングと凝集(アミロイド原性タンパク質)を起こしやすい正常(野生型)タンパク質や変異タンパク質がいくつかあり,そのため,アミロイドーシスには多様な原因と病型が存在する。
アミロイドーシスの発生には,アミロイド原性タンパク質の産生のほかに,おそらくは,こうしたミスフォールディングタンパク質の除去機構の破綻もあると考えられる。アミロイド沈着物は,それ自体は代謝的に不活性であるが,臓器の構造および機能を物理的に妨げる。しかしながら,アミロイド原性タンパク質が線維化する前の段階のオリゴマーには,直接的な細胞毒性を有するものもあり,病態発生機序の重要な構成要素である。
アミロイド沈着物は,ヘマトキシリン-エオジン染色でピンク色に染まり,また,PAS(periodic acid-Schiff)染色またはアルシアンブルー染色で染まる炭水化物成分を含有するが,最大の特徴として,コンゴレッド染色後に偏光顕微鏡下でアップルグリーンの複屈折がみられる。剖検による観察では,罹患臓器は蝋様に見える場合がある。
アミロイドーシスの病因
全身性アミロイドーシスでは,循環血液中のアミロイド原性タンパク質が多様な臓器に沈着物を形成する。全身性アミロイドーシスの主な病型として以下のものがある:
AL(原発性アミロイドーシス):クローン性免疫グロブリン軽鎖の後天性過剰発現が原因である
AF(家族性アミロイドーシス):ミスフォールディングを起こしやすいタンパク質(最も頻度が高いのはトランスサイレチン[TTR])をコードする変異遺伝子の遺伝が原因である
ATTRwt(野生型ATTR;かつては老人性全身性アミロイドーシス[SSA]と呼ばれていた):野生型TTR(wild-type ATTR)のミスフォールディングと凝集が原因である
AA(続発性アミロイドーシス):急性期反応物質である血清アミロイドAの凝集が原因である
β2ミクログロブリンの凝集により引き起こされるアミロイドーシスは,長期間血液透析を行っている患者に生じる場合があるが,発生率は最新の高流量透析膜の使用に伴って低下している。β2ミクログロブリンアミロイドーシスには,該当する遺伝子の変異に起因するまれな遺伝性の病型がある。
限局性アミロイドーシスは,循環血液中のタンパク質ではなく,罹患臓器内で局所的にアミロイド原性タンパク質(ほとんどの場合,免疫グロブリンの軽鎖)が産生されて沈着することで引き起こされると考えられる。頻度が高い発生部位は,中枢神経系(例,アルツハイマー病),皮膚,上気道,下気道,肺実質,膀胱,眼,乳房などである。
ALアミロイドーシス(原発性アミロイドーシス)
ALアミロイドーシスの原因は,単クローン性形質細胞またはその他のB細胞性リンパ増殖性疾患を有する患者において,アミロイド原性の免疫グロブリン軽鎖が過剰産生されることである。軽鎖は,非線維性組織沈着物(すなわち,軽鎖沈着症)を形成することもある。まれに,免疫グロブリンの重鎖がアミロイド線維を形成する(AHアミロイドーシスと呼ばれる)。
アミロイドの一般的な沈着部としては,皮膚,神経,心臓,消化管(舌を含む),腎臓,肝臓,脾臓,血管などがある。通常,骨髄で軽度の形質細胞増加がみられ,多発性骨髄腫に類似するが,真の多発性骨髄腫(溶骨性病変,高カルシウム血症,尿細管円柱,貧血を伴う)を有している患者はほとんどいない。しかし,多発性骨髄腫患者の約10~20%はALアミロイドーシスを発症する。
AFアミロイドーシス(家族性アミロイドーシス)
AFアミロイドーシスの原因は,凝集しやすい変異型の血清タンパク質(通常,肝臓により豊富に産生されるタンパク質)をコードする遺伝子の遺伝である。
AFアミロイドーシスの原因となりうる血清タンパク質としては,トランスサイレチン(TTR),アポリポタンパク質A-I,アポリポタンパク質A-II,リゾチーム,フィブリノーゲン,ゲルソリン,シスタチンCなどがある。最近同定されて家族性と推測されている病型は,血清タンパク質のLECT2(leukocyte chemotactic factor 2)が原因物質であるが,この病型に関わる特定の遺伝子変異の存在は明確には実証されていない。
AFアミロイドーシスのうち最も頻度の高い病型は,ATTRアミロイドーシス(amyloidosis caused by TTR:ATTR)である。TTR遺伝子の130を超える変異とアミロイドーシスとの関連が報告されている。最も頻度の高い変異であるV30Mは,ポルトガル,スウェーデン,ブラジル,および日本でよくみられ,V122I変異はアフリカ系アメリカ人およびカリブ系黒人の約4%にみられる。疾患浸透率と発症年齢には大きな幅がみられるが,家族内および民族集団内では一貫している。ATTRアミロイドーシスは,末梢の感覚神経障害,自律神経性ニューロパチー,慢性腎臓病,および心筋症を引き起こす。一般的に他の神経疾患の症候に先立って手根管症候群が発生する。網膜上皮から変異型TTRが産生されるために硝子体沈着が起きることがあり,また,脈絡叢で変異型TTRが産生されるために軟膜への沈着が起きることもある。
ATTRwtアミロイドーシス(老人性全身性アミロイドーシス)
ATTRwtアミロイドーシスの原因は,臨床的に心臓を標的とする野生型TTRが凝集して沈着することである。
ATTRwtアミロイドーシスは,高齢者における浸潤性心筋症の原因としての認識が高まっている。経カテーテル大動脈弁置換術を受ける大動脈弁狭窄症患者の16%,および駆出率が保持された心不全(HFpEF)で入院した患者の13%にATTRwtアミロイド心筋症が認められる。ATTRwtアミロイドーシスの発生につながる遺伝因子やエピジェネティック因子は不明である。ATTRwtおよびALアミロイドーシスはどちらも心筋症を引き起こすことがあり,また,この年齢層の患者ではアミロイド原性単クローン性免疫グロブリン血症が存在する場合もあるため,ATTRwtアミロイドーシスの患者に誤って化学療法(ALアミロイドーシスに対して使用する)を施行することがないよう,アミロイドの病型を正確に判別することが不可欠である。
AAアミロイドーシス(続発性アミロイドーシス)
この病型は,いくつかの感染症,炎症性疾患,および悪性疾患に続発し,急性期反応物質である血清アミロイドAの各アイソフォームの凝集によって引き起こされる。
一般的に原因となる感染症としては以下のものがある:
素因となる炎症性疾患としては以下のものがある:
これらの疾患で産生されるまたは腫瘍細胞により異所性に分泌される炎症性サイトカイン(例,インターロイキン1[IL-1],腫瘍壊死因子[TNF],IL-6)は,肝臓で血清アミロイドA(SAA)の合成増加を引き起こす。
AAアミロイドーシスの好発部位は,脾臓,肝臓,腎臓,副腎,およびリンパ節である。心臓と末梢または自律神経の障害が疾患経過の後期に生じる。
限局性アミロイドーシス
脳以外での限局性アミロイドーシスは,クローン性免疫グロブリン軽鎖の沈着により起こる場合が最も多く,脳内の限局性アミロイドーシスは,アミロイドβタンパク質により起こる場合が最も多い。
限局性のアミロイド沈着は,典型的には,気道および肺組織,膀胱および尿管,皮膚,乳房,眼を障害する。まれに,局所的に産生されるその他のタンパク質(皮膚に局所的な沈着物を形成するケラチンの各アイソフォームなど)がアミロイドーシスを引き起こす。消化管,気道,および膀胱の粘膜関連リンパ組織で産生されるクローン性免疫グロブリン軽鎖は,これらの臓器における限局性のALアミロイドーシスを引き起こす場合がある。
アミロイドβタンパク質の脳内沈着は,アルツハイマー病または脳アミロイド血管症の一因となる。中枢神経系で産生されるその他のタンパク質が,ミスフォールディング,凝集,およびニューロンの損傷を生じる場合があり,神経変性疾患(例,パーキンソン病,ハンチントン病)を引き起こす。
アミロイドーシスの症状と徴候
全身性アミロイドーシスの症状および徴候は非特異的であり,しばしば診断の遅れにつながる。進行性の多臓器疾患がみられる患者では,アミロイドーシスを強く疑うべきである。
腎臓でのアミロイド沈着は,通常は糸球体膜に生じてタンパク尿を引き起こすが,約15%の症例では尿細管が侵され,微量のタンパク尿を伴う高窒素血症を来す。こうした経過から,顕著な低アルブミン血症,浮腫,および全身浮腫を伴うネフローゼ症候群や,末期腎臓病に進展する場合がある。
肝病変は無痛性の肝腫大を引き起こし,肝腫大は巨大となりうる。肝機能検査では通常,アルカリホスファターゼの上昇とその後ビリルビンの上昇がみられ,肝内胆汁うっ滞が示唆されるが,黄疸はまれである。ときに門脈圧亢進症が生じ,その結果食道静脈瘤および腹水を来す。
気道病変は,呼吸困難,喘鳴,喀血,または気道閉塞を引き起こす。
心筋への浸潤は拘束型心筋症を引き起こし,これにより最終的には拡張機能障害および心不全を来す;心ブロックまたは不整脈が生じることもある。低血圧がよくみられる。
足趾および手指の錯感覚を伴う末梢神経障害は,ALおよびATTRアミロイドーシスの最初の臨床像であることが多い。自律神経性ニューロパチーにより,起立性低血圧,勃起障害,発汗異常,および消化管蠕動障害を来すことがある。
脳アミロイド血管症により自発的な皮質下脳出血を生じることが最も多いが,短い過渡的な神経症状がみられる患者もいる。
消化管アミロイドは小腸および大腸,ならびに食道の蠕動異常を引き起こしうる。胃アトニー,吸収不良,出血,または偽閉塞も生じうる。巨舌症はALアミロイドーシスでよくみられる。
軟部組織のアミロイド病変は,特徴的にATTRwtアミロイドに関連する心筋症の臨床的発現に先行する。軟部組織のアミロイド病変の臨床像としては,手根管症候群,弾発指,上腕二頭筋腱断裂,脊柱管狭窄症などがある。

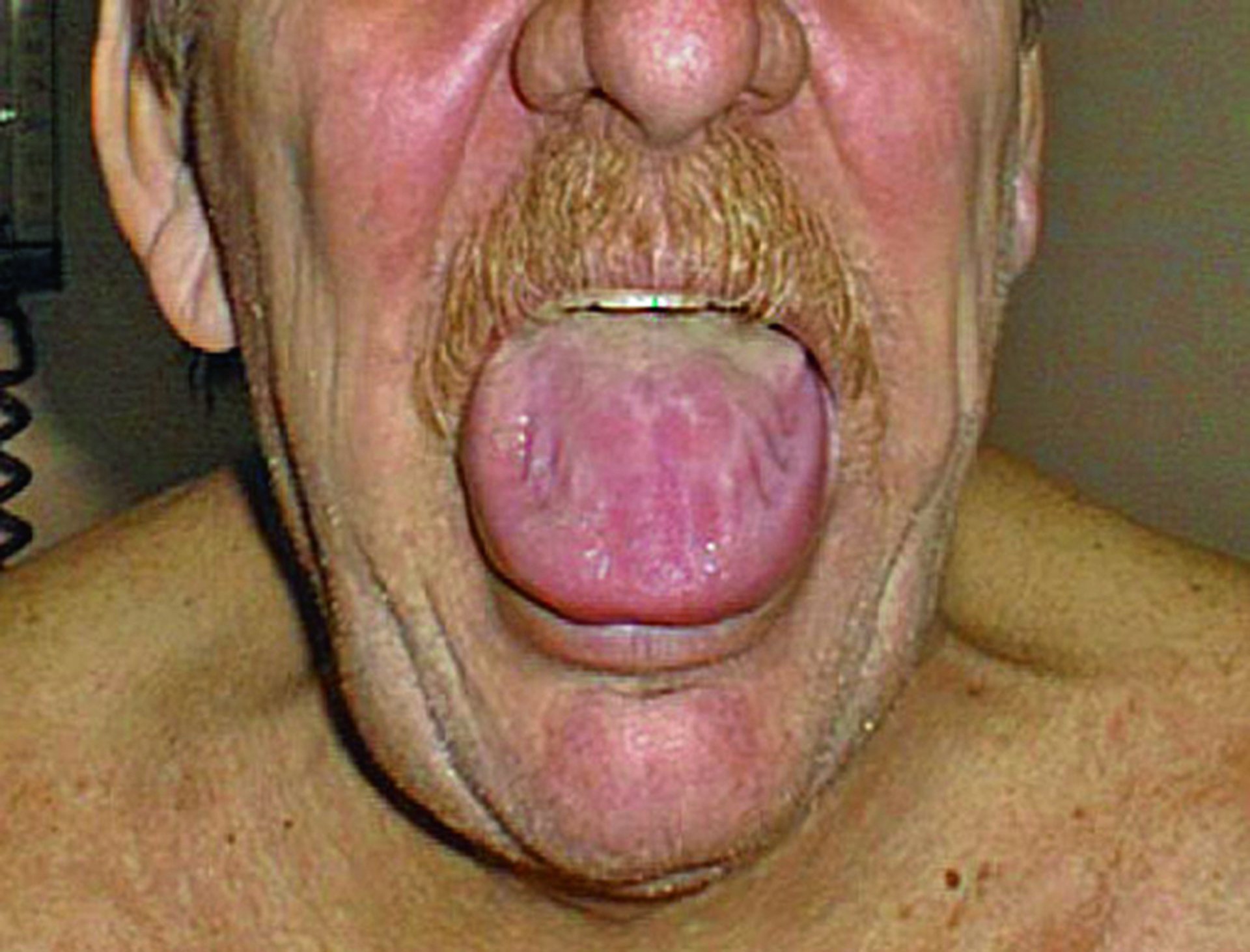
© Springer Science+Business Media
甲状腺アミロイドーシスでは,橋本甲状腺炎に似た,左右対称で圧痛のない硬い甲状腺腫が生じることがある。その他の内分泌障害も生じうる。
肺病変(大半はALアミロイドーシスでみられる)は巣状肺結節,気管気管支病変,またはびまん性肺胞沈着を特徴とする。
アミロイドによる硝子体混濁と両側性の瞳孔縁不整が,いくつかの遺伝性アミロイドーシスで生じる。
皮下出血を含むその他の症状としては,血管へのアミロイド沈着で生じた眼窩周囲の皮下出血(パンダの目徴候[raccoon eyes])などがある。アミロイド沈着により血管が脆弱になり,くしゃみや咳などによる軽微な外傷後に破裂することがある。
アミロイドーシスの診断
生検
アミロイドの病型の判別
臓器病変の検査
生検
アミロイドーシスの診断は,罹患病変における線維性の沈着物を証明することにより行われる。腹部皮下脂肪の吸引は,ALアミロイドーシス患者の約80%,AFアミロイドーシス患者の50%で陽性となるが,ATTRwtアミロイドーシス患者では陽性となるのは25%未満である。脂肪生検の結果が陰性であった場合は,臨床的に障害されている臓器の生検を行うべきである。腎生検および心生検の診断感度は,これらの臓器が臨床的に障害されている場合はほぼ100%である。組織切片をコンゴレッドで染色して,偏光顕微鏡で特徴的な複屈折を観察する。心臓または腎臓の生検検体を電子顕微鏡下で観察すると,10nmの枝分かれのない線維も確認できる。
アミロイドの病型の判別
生検によりアミロイドーシスを確認したら,種々の手法を用いてその病型を判別する。アミロイドーシスの一部の病型には,免疫組織化学法または蛍光抗体法が診断に有用なことがあるが,偽陽性が生じることもある。その他の有用な手法には,遺伝子配列決定(AFアミロイドーシスに対して)および質量分析法(感度および特異度が最も高い方法)による生化学的同定などがある。
ALアミロイドーシスが疑われた場合は,基礎に形質細胞疾患がないか評価すべきであり,方法としては血清遊離免疫グロブリン軽鎖の定量,免疫固定電気泳動を用いた血清中または尿中単クローン性軽鎖の定性的検出(血清タンパク質電気泳動および尿タンパク質電気泳動は,ALアミロイドーシスを有する患者では感度が低い),および骨髄生検とフローサイトメトリーまたは免疫組織化学的検査を用いて,形質細胞がクローン性に増殖していることを証明すべきである。
クローン性の形質細胞が10%を超える患者では,溶骨性病変,貧血,腎機能不全,および高カルシウム血症のスクリーニングなどを行い,多発性骨髄腫の診断基準を満たすかどうか確認すべきである。
臓器病変
まず非侵襲的検査で臓器障害のスクリーニングを行う:
腎臓:尿検査,血清中BUN,およびクレアチニンの測定
肝臓:肝機能検査
肺:胸部X線,胸部CT,および/または肺機能検査
心臓:心電図検査および脳(B型)ナトリウム利尿ペプチド(BNP)またはN-terminal-pro BNP(NT-pro-BNP)およびトロポニンなどのバイオマーカーの測定
心電図上で低電位(心室の肥厚により引き起こされる)および/またはリズム異常を認めれば,心障害が示唆される可能性がある。症状,心電図,または心筋バイオマーカーから心障害が示唆される場合は,心エコー検査を行い,拡張期の弛緩と収縮機能を測定し,両室肥大のスクリーニングを行う。不明瞭な症例では,心臓MRIを施行し,特徴的な所見である心内膜下のガドリニウム遅延造影効果を検出する。心臓のテクネチウムピロリン酸による核医学検査の進歩により,ATTRアミロイドに関連する心疾患の検出が改善され,血液検査でALアミロイドーシスが除外されれば,心臓生検の必要性を回避できる(1, 2)。
診断に関する参考文献
1.Gillmore JD, Maurer MS, Falk RH, et al: Nonbiopsy diagnosis of cardiac transthyretin amyloidosis.Circulation 133(24):2404–2412, 2016.
2.Maurer MS, Bokhari S, Damy T, et al: Expert consensus recommendations for the suspicion and diagnosis of transthyretin cardiac amyloidosis.Circ Heart Fail 12(9):e006075, 2019.
アミロイドーシスの予後
予後はアミロイドーシスの病型および侵された器官系により異なるが,疾患に応じた適切な治療および支持療法を行うことにより,多くの患者で非常に良好な期待余命が得られる。
重度の心筋症を合併したALアミロイドーシスは予後が最も不良であり,生存期間の中央値は1年未満である。未治療のATTRアミロイドーシスは,通常は5~15年以内に末期の心疾患または神経疾患に進展する。ATTRwtアミロイドーシスは,かつては心臓を侵す全身性アミロイドーシスの中で最も進行が遅い病態と考えられていたが,ATTRwtアミロイドーシスの患者には生検による診断から中央値4年以内に症候性の心不全に進行して死亡する者もいる。
AAアミロイドーシスの予後は,基礎にある感染症,炎症,または悪性疾患に対する治療の有効性に大きく依存する。
アミロイドーシスの治療
支持療法
病型特異的な治療
現在,アミロイドーシスの大半の病型に対する特異的治療法が存在するものの,一部の治療法は研究中である。全身性アミロイドーシスのいずれの病型に対しても,支持療法の施行が症状の緩和および生活の質の向上に役立つ可能性がある。
支持療法
支持療法は侵された器官系を対象とする:
腎臓:ネフローゼ症候群と浮腫がみられる患者は,塩分・水分制限,およびループ利尿薬により治療すべきであり,持続的なタンパク質喪失があるため,タンパク質の摂取は制限すべきではない。腎移植は,基礎疾患の経過がコントロールされているときの1つの選択肢であり,他の腎疾患と比べて長期の生存期間を得ることが可能である。
心臓:心筋症がみられる患者は,塩分・水分制限とループ利尿薬により治療すべきである。心不全に対するその他の治療薬(ジゴキシン,アンジオテンシン変換酵素(ACE)阻害薬,カルシウム拮抗薬,β遮断薬など)は,忍容性が不良であるため禁忌である。重度の心病変を有するALまたはATTRアミロイドーシス患者のうち慎重に選択された症例では,心臓移植が成功を収めている。移植された心臓での再発を予防するため,ALアミロイドーシス患者にはクローン性形質細胞疾患を標的とする積極的な化学療法を施行する必要があり,症状を伴うATTRアミロイドポリニューロパチーまたは心筋症の患者には抗TTR療法を考慮すべきである。
消化管:下痢がみられる患者にはロペラミドが有益となる場合がある。早期満腹感および胃内容物貯留がみられる患者には,メトクロプラミドが有益な場合がある。
神経系:末梢神経障害がみられる患者は,ガバペンチン,プレガバリン,またはデュロキセチンにより疼痛が緩和される場合がある。
起立性低血圧は,高用量ミドドリンにより改善する場合が多い;ミドドリンは高齢男性で尿閉を引き起こす場合があるが,この年齢の男性で本薬剤の合併症である臥位高血圧が問題となることはまれである。サポートストッキングが有用な場合があり,末梢浮腫,全身浮腫,または心不全がみられない患者では,フルドロコルチゾンを使用できる。
ALアミロイドーシス
ALアミロイドーシスに対しては:
臓器機能を温存し,余命を延長するため,形質細胞に対する治療を速やかに開始することが不可欠である。
多発性骨髄腫に対して使用される薬剤の大半が,ALアミロイドーシスに対して使用されている;臓器機能が損なわれた場合は,しばしば,薬剤,用量,およびスケジュールの選択を変更しなければならない。
アルキル化薬(例,メルファラン,シクロホスファミド)とコルチコステロイドの併用による化学療法は,便益が示された最初のレジメンであった。高用量のメルファランの静脈内投与と自家造血幹細胞移植の併用は,選択された患者において非常に効果的となる場合がある(1)。
プロテアソーム阻害薬(例,ボルテゾミブ)および免疫調節薬(例,レナリドミド)も効果的となる可能性がある。併用療法およびsequential療法が現在研究中である。
形質細胞は放射線に対する感受性が非常に高いため,限局性のALアミロイドーシスは低線量の外照射療法により治療できる。
ATTRアミロイドーシス
ATTRアミロイドーシスに対しては:
肝移植
四量体安定化薬
遺伝子サイレンシング薬
肝移植(変異タンパク質合成部位を除去し,正常なTTR産生部位を移植する)は,発症時(初期の神経障害があり心病変を伴わない)に行う場合,特定のTTR変異に対し効果的となる場合がある。疾患後期,すなわち心臓および神経に有意なアミロイド沈着が生じた後に肝移植を施行した場合,野生型TTRのミスフォールディングおよび既存のアミロイド沈着部位への沈着により,アミロイドに関連する心筋症および神経障害の進行にしばしばつながる。
いくつかの薬剤の中には,血漿内に循環しているTTR四量体を安定化させ,TTRのミスフォールディングおよび線維形成を予防し,生活の質を維持しつつ効果的に神経疾患の進行を抑制することが証明されているものもある。こうしたTTR安定化薬としては,広く入手可能なジェネリックの抗炎症薬であるジフルニサルや,タファミジスなどがある(2, 3)。
アンチセンスRNAまたはRNA干渉を用いてTTR mRNAの翻訳を阻害するTTR遺伝子サイレンシングは,TTRの血清中濃度を効率的に抑制し,約70%の患者で神経学的予後を改善させ,一部の患者では損傷した神経を修復できると考えられる(4, 5)。2つの遺伝子サイレンシング薬であるパチシランおよびイノテルセン(inotersen)が利用可能でる。
ATTRwtアミロイドーシス
ATTRwtアミロイドーシスに対しては:
四量体安定化薬
ATTRアミロイド心筋症患者において,タファミジスを使用したTTRの安定化は,全死亡率および心血管関連の入院を減少させることが示されている(3)。
遺伝性ATTRアミロイドーシスとは異なり,ATTRwtアミロイドーシスの患者では,野生型のアミロイド原性タンパク質が構造的に正常なTTRであるため,肝移植は効果的ではない。
AAアミロイドーシス
家族性地中海熱により引き起こされるAAアミロイドーシスでは,コルヒチン0.6mg,経口,1日1回または1日2回の投与が効果的である。
AAアミロイドーシスのその他の病型には,基礎にある感染症,炎症性疾患,または悪性腫瘍を標的とした治療を行う。
サイトカインのシグナル伝達を阻害するためにIL-1,IL-6,およびTNF阻害薬が用いられることがあり,これによって肝臓における血清アミロイドA(SAA)の産生を促進する炎症過程を軽減することができる。
治療に関する参考文献
1.Sanchorawala V, Sun F, Quillen K, et al: Long-term outcome of patients with AL amyloidosis treated with high-dose melphalan and stem cell transplantation: 20-year experience.Blood 126: 2345–2347, 2015. doi: 10.1182/blood-2015-08-662726.Epub 2015 Oct 6
2.Berk JL, Suhr OB, Obici L, et al: Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial.JAMA 310: 2658–2667, 2013. doi: 10.1001/jama.2013.283815
3.Maurer MS, Schwart JH, Gundapaneni B, et al: Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy.N Engl J Med 379:1007–1016, 2018.
4.Adams D, Gonzalez-Duarte A, O'Riordan WD, et al: Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis.N Engl J Med 379:11–21, 2018.
5.Benson MD, Waddington-Cruz M, Berk JL, et al: Inotersen treatment for patients with transthyretin amyloidosis.N Engl J Med 379:22–31, 2018.
アミロイドーシスの要点
アミロイドーシスは,特定のミスフォールディングタンパク質が凝集して不溶性の線維となり,それらが臓器に沈着して機能障害を引き起こす疾患群である。
ミスフォールディングを起こしやすいタンパク質には多数の様々な種類がある;こうしたタンパク質は,遺伝子異常または特定の病態により産生されるタンパク質であったり,単クローン性形質細胞またはその他のB細胞性リンパ増殖性疾患により産生される免疫グロブリン軽鎖であったりする。
アミロイド原性タンパク質によりアミロイドの病型および疾患の臨床経過が決まるが,異なる病型の臨床像が重複する場合もある。
多数の臓器が侵される可能性があるが,心障害は特に予後不良であり,アミロイド心筋症を発症すると,典型的には拡張機能障害,心不全,ならびに心ブロックおよび/または不整脈に至る。
診断には生検による;アミロイドーシスの病型は,種々の免疫学的検査,遺伝子検査,および生化学的検査により判別される。質量分析法はアミロイドの病型の判別において感度および特異度が最も高い方法である。
適切な支持療法は症状の緩和と生活の質の向上に役立ち,選択された患者には臓器移植が助けになる可能性がある。
基礎疾患の治療を行う;形質細胞またはリンパ球増殖性疾患によるALアミロイドーシスでは,化学療法が非常に効果的となる場合がある;続発性AAアミロイドーシスでは,抗感染症薬および抗炎症薬が役立つ可能性がある。
遺伝性ATTRアミロイドーシスでは,低分子安定化薬による治療および遺伝子サイレンシング薬により,神経機能の悪化が抑制されるか,改善する可能性がある;アミロイド心筋症患者(ATTRまたはATTRwt)では,タファミジスにより全死亡率および心血管関連の入院が減少する。







