




21-水酸化酵素(CYP21A2)欠損により,副腎のホルモン前駆体がコルチゾールへ,また一部の症例ではアルドステロンへ適切に変換されず,その結果ときに重度の低ナトリウム血症および高カリウム血症が引き起こされる。蓄積されたホルモン前駆体はアンドロゲン産生経路へ流入し,男性化を引き起こす。診断はコルチゾール,その前駆体,副腎アンドロゲンの値を,ときに副腎皮質刺激ホルモン投与後に測定することによる。治療はグルココルチコイドおよび,必要に応じて,ミネラルコルチコイドにより行い,性別不明性器がみられる一部の新生児女児には外科的再建を行う。
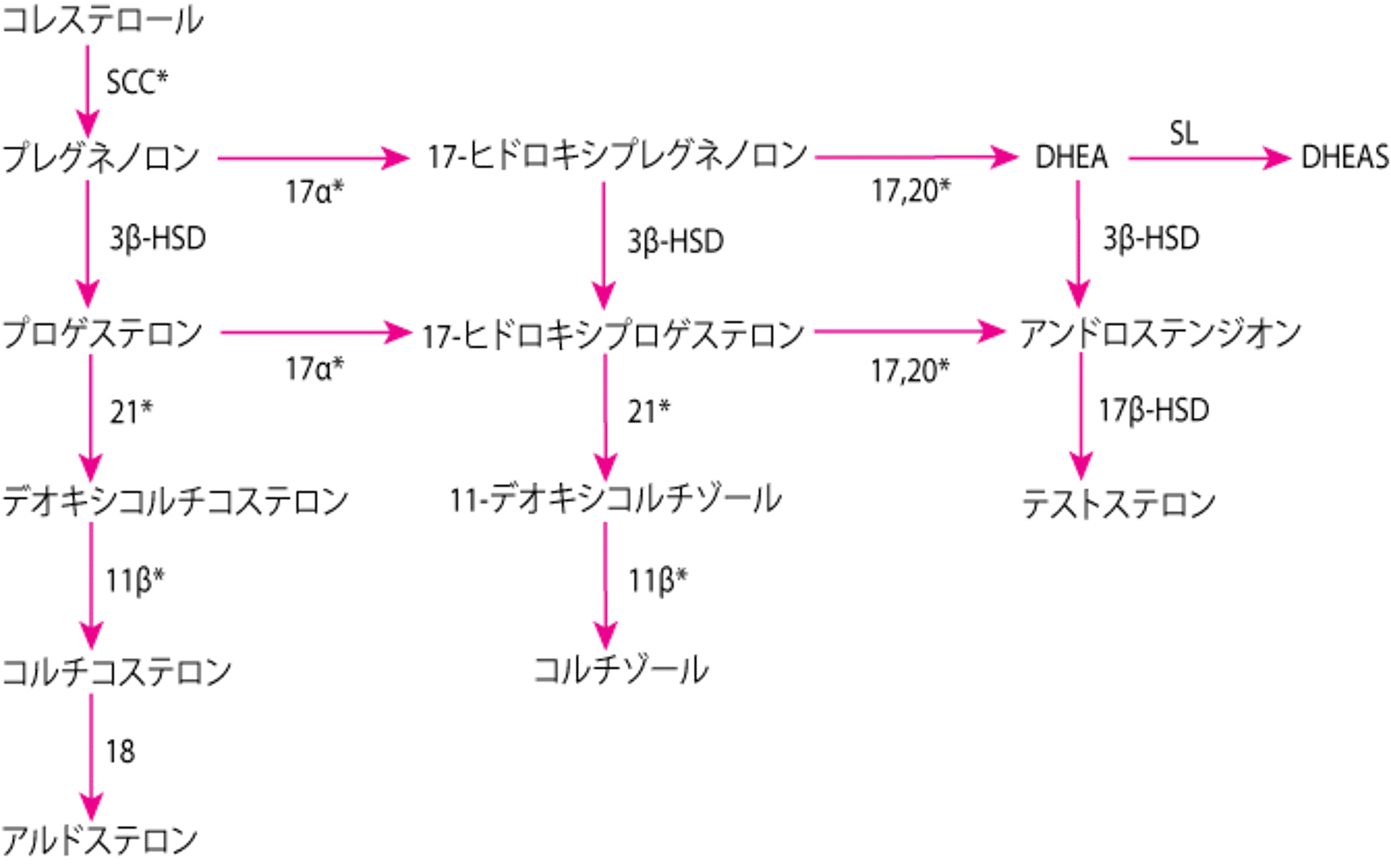
先天性副腎過形成症の全症例の90%は,21-水酸化酵素欠損症が原因である。発生率は出生10,000~15,000例当たり1例である。疾患の重症度は具体的なCYP21A2変異および酵素欠損の程度により決まる。酵素欠損により,17-ヒドロキシプロゲステロンからコルチゾール前駆体である11-デオキシコルチゾールへの変換,およびプロゲステロンからアルドステロン前駆体であるデオキシコルチコステロンへの変換が完全または部分的に遮断される。コルチゾール合成が減少するため,副腎皮質刺激ホルモン(ACTH)値が上昇し,これが副腎皮質を刺激し,コルチゾール前駆体(例,17-ヒドロキシプロゲステロン)の蓄積および副腎アンドロゲンデヒドロエピアンドロステロン(DHEA)およびアンドロステンジオンの過剰産生が起こる。アルドステロン欠乏によって,塩喪失,低ナトリウム血症,および高カリウム血症が生じうる(1, 2)。
副腎ホルモンの合成
*副腎皮質刺激ホルモン(ACTH)によって刺激される酵素 |
11β = 11β-水酸化酵素(P-450c11);17α = 17α-水酸化酵素(P-450c17);17,20 = 17,20リアーゼ(P-450c17);18 = アルドステロン合成酵素(P-450aldo);21 = 21-水酸化酵素(P-450c21);DHEA = デヒドロエピアンドロステロン;DHEAS = DHEA硫酸エステル;3β-HSD = 3β-ヒドロキシステロイド脱水素酵素(3β2-HSD); 17β-HSD = 17β-ヒドロキシステロイド脱水素酵素(17β-HSD);SCC = 側鎖切断(P-450scc);SL = スルホトランスフェラーゼ(SULT1A1,SULT1E1)。 |

古典型21-水酸化酵素欠損症
古典型21-水酸化酵素欠損症は2型に分けられる:
塩喪失型
単純男性化型
両型とも,副腎アンドロゲン値が上昇し男性化が起こる。
塩喪失型は最重症であり古典型21-水酸化酵素欠損症例の70%を占める;酵素活性の完全欠損が認められ,コルチゾールおよびアルドステロンが非常に低値となる。アルドステロンの分泌が最小限となり,塩が失われ,低ナトリウム血症,高カリウム血症,および血漿レニン活性上昇が生じる。
単純男性化型では,コルチゾール合成が障害され,アンドロゲン活性が上昇するが,アルドステロン産生を正常,または軽度のみの低下に維持する十分な酵素活性がある。
非古典型21-水酸化酵素欠損症
非古典型21-水酸化酵素欠損症は,古典型21-水酸化酵素欠損症より一般的である。発生率には白人集団での出生1000~2000件当たり1例(0.1~0.2%)から特定の民族集団(例,アシュケナージ系ユダヤ人)での1~2%までの幅がある。非古典型21-水酸化酵素欠損症では20~50%の21-水酸化酵素活性があり,重症度のより低い病態が起こる(対して古典型21-水酸化酵素欠損症では0~5%の酵素活性)。アルドステロンおよびコルチゾール値は正常なため塩喪失はないが,副腎アンドロゲン値が軽度上昇しているため,小児期または成人期で軽度のアンドロゲン過剰が起こる。
総論の参考文献
1.Witchel SF: Congenital adrenal hyperplasia.J Pediatr Adolesc Gynecol 30(5):520–534, 2017. doi: 10.1016/j.jpag.2017.04.001
2.El-Maouche D, Arlt W, Merke DP: Congenital adrenal hyperplasia.Lancet 390(10108):2194–2210, 2017. doi: 10.1016/S0140-6736(17)31431-9
症状と徴候
塩喪失型は,男性化に加えて低ナトリウム血症(ときに重度),高カリウム血症,低血圧の原因となる。診断および治療がなされなければ,嘔吐,下痢,低血糖,循環血液量減少,およびショックを伴った,生命を脅かす副腎クリーゼへとつながる可能性がある。
古典型21-水酸化酵素欠損症のいずれの型でも,新生児女児には性別不明の外性器がみられ,陰核肥大,大陰唇の癒合,尿生殖洞(尿道や腟の明確な開口部ではない)を伴う。男児では一般に正常な外性器発育を認めるため,塩喪失型の診断が遅れることがある;罹患男児はルーチンの新生児スクリーニングによってのみ同定されることが多い。新生児スクリーニングにより発見されない場合,単純男性化型の男児は,アンドロゲン過剰徴候が発生するまで数年間診断されない可能性がある。アンドロゲン過剰の徴候には,早期の陰毛の出現,男女ともに成長速度の増加,女児では陰核肥大,男児では陰茎肥大およびより早期の声の低音化などが挙げられる。
非古典型21-水酸化酵素欠損症の小児は出生時に症状がなく,通常,小児期または青年期まで症状を認めない。罹患女児では早期の陰毛発育,骨年齢の進行,多毛,希発月経,ざ瘡などがみられ,これらの症状が多嚢胞性卵巣症候群の臨床像に類似することがある。罹患男児では早期の陰毛の発生,成長加速,および骨年齢の進行が認められる。
罹患した女児で,特に塩喪失型の場合,成人に達すると生殖機能が障害されることがある;陰唇癒合および無排卵周期または無月経のことがある。塩喪失型の男児では,成人になって生殖能力がある者もいるが,精巣の副腎遺残腫瘍(慢性的なACTHによる刺激下で肥大した副腎組織から成る良性精巣内腫瘤),ライディッヒ細胞機能不全,テストステロン減少,および精子形成障害を起こす者もいる。塩喪失型以外の型に罹患した男児の大部分は,治療されなくても生殖能力はあるが,一部において精子形成が障害される。
診断
血液検査
ときにACTH刺激試験
ときに遺伝子型解析
ルーチンの新生児スクリーニングには,典型的には血清17-ヒドロキシプロゲステロン値測定が含まれる。この値が上昇している場合,血中コルチゾール低値の同定,ならびに血中のDHEA,アンドロステンジオン,およびテストステロン高値の同定により21-水酸化酵素欠損症の診断が確定する。まれに診断が不明確となり,ACTH投与前と投与60分後にこれらのホルモンのレベルを測定しなければならないことがある(ACTHまたはテトラコサクチド刺激試験)。後に症状が出現する患者においてはACTH刺激試験が役立つことがあるが,遺伝子型解析が必要になる可能性もある。
塩喪失型の小児では低ナトリウム血症および高カリウム血症が認められる;デオキシコルチコステロン,コルチコステロン,およびアルドステロンが低値であり,レニンが高値である。
出生前のスクリーニングおよび診断(および実験的治療)が可能であり,高リスクならば(例,遺伝性欠損があり罹患した同胞がいる胎児),CYP21遺伝子の分析を行う。保因者の状態(ヘテロ接合体性)は小児および成人で決定されうる。
治療
コルチコステロイドの補充
ミネラルコルチコイドの補充(塩喪失型)
ときに外科的再建術
乳児の副腎クリーゼには,輸液による緊急治療が必要となる。塩喪失型が疑われる場合,副腎クリーゼを予防するため,負荷量のヒドロコルチゾン(100mg/m2/日)の持続静注を行う;投与量は数週間かけて,より生理学的な補充の量まで減少させる。
維持療法は,不足したステロイドの補充としてのコルチコステロイドである(典型的には,経口でヒドロコルチゾン3.5~5mg/m2,1日3回,1日総量として通常は20mg/m2以下)。思春期後の青年および成人は,プレドニゾン5~7.5mgの1日1回経口投与もしくは2.5~3.75mgの1日2回投与,またはデキサメタゾン0.25~0.5mgの1日1回投与もしくは0.125~0.25mgの1日2回投与により治療される。
治療への反応を乳児では3カ月毎に,生後12カ月を超える小児では3~4カ月毎にモニタリングする。コルチコステロイドによる過剰治療は,医原性クッシング症候群をもたらす結果となり,肥満,正常以下の発育,骨成熟の遅滞を引き起こす。過少な治療は,結果としてACTH抑制の不能を招き,小児において男性化および正常を上回る成長速度の原因となって,最終的には成長の早期終了および低身長をもたらす。モニタリングとして,血清中の17-ヒドロキシプロゲステロン,アンドロステンジオン,およびテストステロンを測定するとともに,毎年成長速度と骨成熟を評価する。
塩喪失型の維持療法は,コルチコステロイドに加えて,ナトリウムおよびカリウムの恒常性回復のためのミネラルコルチコイドの補充である。経口フルドロコルチゾン(通常は0.1mg,1日1回,範囲は0.05~0.3mg)が塩喪失がある場合に投与される。乳児はしばしば経口による塩分補給を約1年間必要とする。治療中の綿密なモニタリングが不可欠である。
病気になったときは,副腎クリーゼを予防するため,コルチコステロイドの用量を増量する(一般的には2倍または3倍)。ミネラルコルチコイドの補充量は変更しない。経口治療で不安がある場合(例,重度の嘔吐または生命を脅かす状況),ヒドロコルチゾン(50~100mg/m2)を筋注で単回投与できる。注射した場合,一般的には患児を救急外来で評価し,輸液,コルチコステロイドの追加,またはその両方が必要であるかどうかを判断する必要がある。
罹患した女子乳児は,陰核形成の整復と腟開口部の造設による外科的再建を必要とすることがある。しばしば,成人期にさらなる手術が必要となる。性心理の問題に対する適切なケアと配慮があれば,正常な性生活と妊孕性を期待できる。
出生前の治療では,胎児の下垂体からのACTH分泌を抑制するために,コルチコステロイド(通常デキサメタゾン)を母親に投与する;これにより罹患した胎児女児の男性化の程度を軽減するか,または防ぐことが可能となる。治療は,実験的なものにはなるが,懐胎した最初の数週間のうちに開始する必要がある。
非古典型21-水酸化酵素欠損症の治療は症状によって異なる。無症状の場合,治療の必要はない。症状がある場合,コルチコステロイド治療は古典型21-水酸化酵素欠損症と同様であるが,より低用量で効果的となることが多い。ミネラルコルチコイドの補充は必要ではない。
要点
21-水酸化酵素欠損症の小児では様々な程度のアンドロゲン過剰が認められ,約70%にアルドステロン欠乏による塩喪失型が認められる。
女児では,通常は出生時にアンドロゲン過剰が性別不明の外性器(例,陰核肥大,大陰唇癒合,尿道や腟の明確な開口部でない尿生殖洞)として現れる;その後,多毛,希発月経,およびざ瘡を呈する可能性がある。
男児では,アンドロゲン過剰は明白でないか,または小児期に成長速度の上昇および思春期の早発徴候を現すことがある。
両性で,塩喪失により低ナトリウム血症および高カリウム血症が起こる。
ステロイドホルモンならびにときに副腎皮質刺激ホルモン(ACTH)刺激および/または遺伝子型解析により診断する。
コルチコステロイドおよびときにミネラルコルチコイドの補充により治療する;女児では外科的再建術を必要とすることがある。







