


Le cardiopatie congenite sono le anomalie congenite più comuni, verificandosi in quasi l'1% dei nati vivi (1). Tra i difetti di nascita, la cardiopatia congenita è la principale causa di mortalità infantile.
Le più comuni malattie cardiache congenite diagnosticate nell'infanzia sono i difetti del setto interventricolare (comunicazione interventricolare) muscolare e perimembranoso seguiti dai difetti del setto interatriale tipo ostium secondum, con una prevalenza totale di 48,4 su 10 000 nati vivi (2, 3, 4). La più comune cardiopatia congenita cianotica è la tetralogia di Fallot, che è due volte più prevalente della trasposizione dei grossi vasi (4,7 vs 2,3/10 000 nascite). Complessivamente, le valvole aortiche bicuspidi sono i difetti congeniti più frequenti con una prevalenza riportata dallo 0,5% al 2,0%.
Riferimenti generali
1. Reller MD, Strickland MJ, Riehle-Colarusso T, et al: Prevalence of congenital heart defects in metropolitan Atlanta, 1998–2005. J Pediatr 153(6):807–813, 2008.
2. Freeze SL, Landis BJ, Ware SM, Helm BM: Bicuspid aortic valve: a review with recommendations for genetic counseling. J Genet Couns 25(6):1171–1178, 2016.
3. van der Linde D, Konings EEM, Slager MA, et al: Birth prevalence of congenital heart disease worldwide: a systematic review and meta-analysis. J Am Coll Cardiol 58(21):2241–2247, 2011. doi: 10.1016/j.jacc.2011.08.025
4. Daubeney PEF, Rigby ML, Niwa K, Gatzoulis MA (eds): Pediatric Heart Disease: A Practical Guide. Wiley-Blackwell 2012.
Eziologia della cardiopatia congenita
Complessi fattori ambientali e genetici contribuiscono allo sviluppo delle cardiopatie congenite.
Fattori ambientali comuni includono malattie materne (p. es., diabete, rosolia, lupus eritematoso sistemico) o l'assunzione materna di agenti teratogeni (p. es., litio, isotretinoina, anticonvulsivanti). L'età materna è un noto fattore di rischio per alcune condizioni genetiche, in particolare la Sindrome di Down, che può comprendere difetti cardiaci. Non è chiaro se l'età materna sia un fattore di rischio indipendente per la cardiopatia congenita. Anche l'età paterna può essere un fattore di rischio (1).
Alcune anomalie cromosomiche numeriche (aneuploidie), come per esempio la trisomia 21 (sindrome di Down), la trisomia 18, la trisomia 13 e la monosomia X (sindrome di Turner), sono fortemente associate a cardiopatie congenite. Tuttavia, queste anomalie rappresentano solo il 5-6% dei pazienti con cardiopatie congenite.
Molti altri casi riguardano delezioni subcromosomiche (microdelezioni), duplicazioni subcromosomiche, o mutazioni di un singolo gene. Spesso queste mutazioni causano sindromi congenite che interessano più organi oltre al cuore. Alcuni esempi comprendono la sindrome di DiGeorge (microdelezione in 22q11.2) e la sindrome di Williams (talvolta conosciuta come sindrome di Williams-Beuren) (microdelezione in 7p11.23). Difetti di un singolo gene che causano sindromi associate a cardiopatie congenite sono le mutazioni di fibrillina-1 (sindrome di Marfan), TXB5 (sindrome di Holt-Oram), e PTPN11 (sindrome di Noonan). I difetti di un singolo gene possono anche causare isolati difetti cardiaci congeniti (ossia, non sindromici).
Non viene rilevata alcuna eziologia genetica identificabile in circa il 72% dei pazienti con cardiopatia congenita (2, 3, 4).
Il rischio di recidiva delle cardiopatie congenite in una famiglia varia a seconda della causa. Il rischio è trascurabile nelle mutazioni de novo, dal 2 al 5% nelle cardiopatie congenite multifattoriali non sindromiche e del 50% quando la causa è una mutazione autosomica dominante. L'identificazione di una valvola aortica bicuspide in un individuo merita uno screening familiare in vista della prevalenza familiare riportata del 9% (5). È importante identificare i fattori genetici perché più pazienti con cardiopatie congenite, rispetto al passato, sono in grado di sopravvivere fino all'età adulta e potenzialmente mettono su famiglia.
Riferimenti relativi all'eziologia
1. Materna-Kiryluk A, Wiśniewska K, Badura-Stronka M, et al: Parental age as a risk factor for isolated congenital malformations in a Polish population. Paediatr Perinat Epidemiol 23(1):29-40, 2009. doi: 10.1111/j.1365-3016. 2008.00979.x
2. Russell MW, Chung WK, Kaltman JR, Miller TA: Advances in the understanding of the genetic determinants of congenital heart disease and their impact on clinical outcomes. J Am Heart Assoc 7(6):e006906, 2018. doi:10.1161/JAHA.117.006906
3. van der Linde D, Konings EEM, Slager MA, et al: Birth prevalence of congenital heart disease worldwide: a systematic review and meta-analysis. J Am Coll Cardiol 58(21):2241–2247, 2011. doi: 10.1016/j.jacc.2011.08.025
4. Pierpont ME, Brueckner M, Chung WK, et al: Genetic Basis for Congenital Heart Disease: Revisited: A Scientific Statement From the American Heart Association [published correction appears in Circulation 2018 Nov 20;138(21):e713]. Circulation 138(21):e653–e711, 2018. doi:10.1161/CIR.0000000000000606
5. Freeze SL, Landis BJ, Ware SM, Helm BM: Bicuspid aortic valve: a review with recommendations for genetic counseling. J Genet Couns 25(6):1171–1178, 2016.
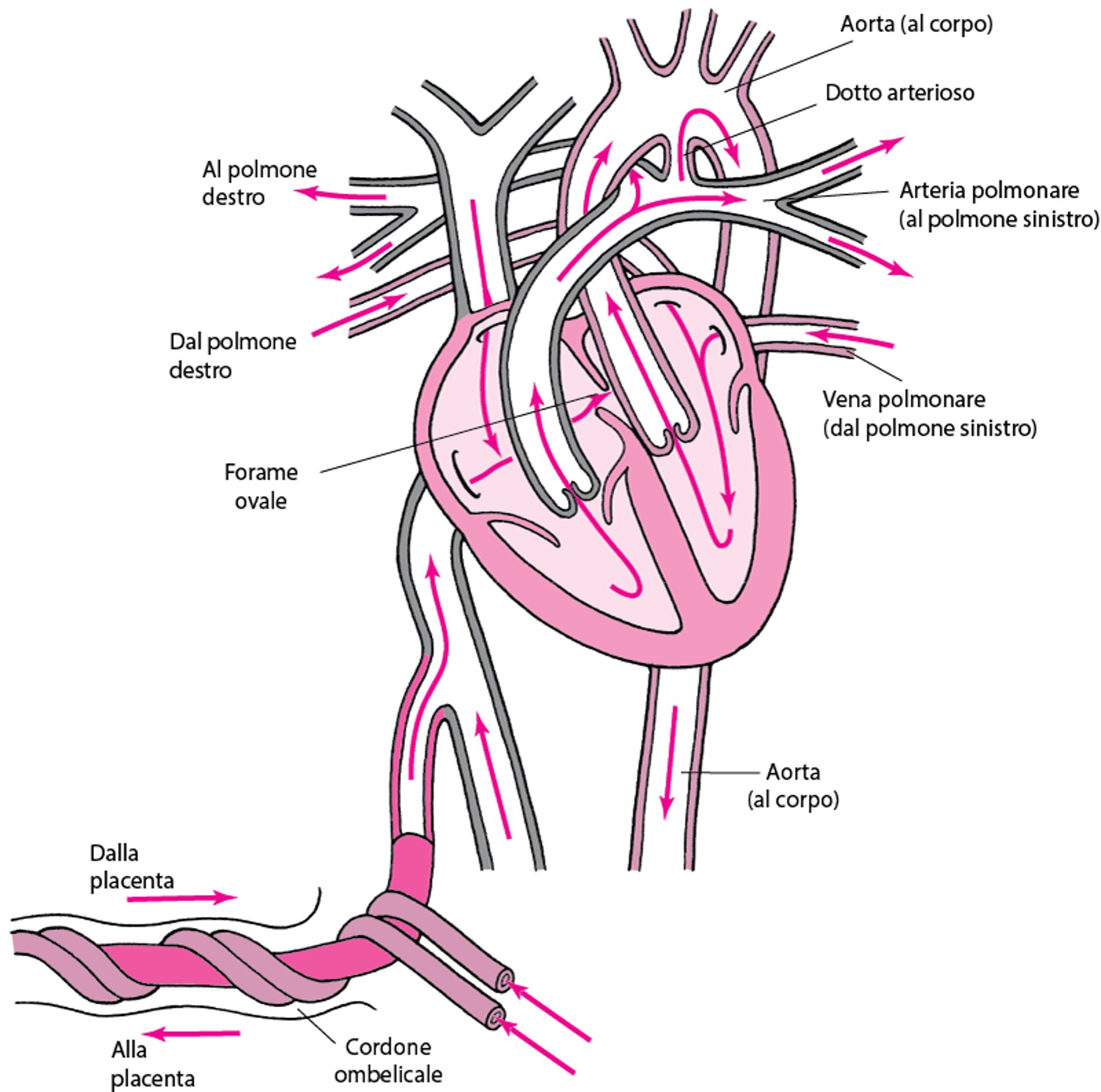
Circolazione fetale normale
La circolazione fetale è caratterizzata da
Uno shunt destro-sinistro di sangue attorno ai polmoni non ventilati attraverso il dotto arterioso pervio (che connette l'arteria polmonare all'aorta) e il forame ovale (che mette in comunicazione l'atrio destro e quello sinistro)
Lo shunt è favorito dalle alte resistenze arteriolari polmonari e dalla resistenza al flusso ematico relativamente bassa nel circolo sistemico (placenta inclusa). Circa il 90-95% del flusso in uscita dal cuore destro bypassa i polmoni e raggiunge direttamente la circolazione sistemica. Il dotto arterioso fetale è mantenuto aperto dalla PaO2 sistemica fetale bassa (circa 25 mmHg) insieme alle prostaglandine prodotte localmente. Il forame ovale è mantenuto aperto dai gradienti delle pressioni atriali: nel feto la pressione atriale sinistra è relativamente bassa a causa del ridotto ritorno ematico polmonare, ma la pressione atriale destra è relativamente alta per il notevole ritorno ematico dalla placenta.
Circolazione normale in un feto
Nel feto, il sangue che entra nel lato destro del cuore è già stato ossigenato attraverso la placenta. Poiché i polmoni non sono ventilati, solo una piccola quantità di sangue deve passare attraverso l'arteria polmonare. La maggior parte del sangue dal lato destro del cuore bypassa i polmoni attraverso il
Normalmente queste due strutture si chiudono poco dopo la nascita. |

Cambiamenti perinatali
Dopo i primi atti respiratori, si verificano profonde modificazioni a livello della circolazione sistemica
Aumento del flusso sanguigno polmonare
Chiusura funzionale del forame ovale
Le resistenze arteriolari polmonari cadono improvvisamente come risultato della vasodilatazione derivante dall'espansione polmonare, dall'aumentata PaO2 e dalla ridotta PaCO2. Le forze elastiche delle coste e della parete toracica diminuiscono la pressione interstiziale polmonare, determinando un ulteriore aumento del flusso ematico attraverso i capillari polmonari. L'aumentato ritorno venoso dai polmoni incrementa la pressione atriale sinistra, riducendo così la pressione differenziale tra atrio sinistro e destro; questo effetto contribuisce alla chiusura funzionale del forame ovale.
Appena il flusso polmonare si stabilizza, il ritorno venoso polmonare aumenta e la pressione atriale sinistra s'innalza. L'introduzione d'aria incrementa la PaO2, cosa che determina la costrizione delle arterie ombelicali. Il flusso ematico placentare si riduce o cessa del tutto e si riduce il ritorno ematico all'atrio destro. Così, la pressione atriale destra diminuisce mentre aumenta la pressione atriale sinistra; di conseguenza, i 2 componenti fetali del setto interatriale (septum primum e septum secundum) vengono accostati, arrestando il flusso attraverso il forame ovale. Nella maggior parte delle persone, i 2 setti alla fine si fondono e il forame ovale cessa di esistere. Tuttavia, nel 25% degli adulti, il forame ovale può rimanere pervio con uno shunt residuo minimo o nullo (1).
Subito dopo la nascita, le resistenze sistemiche superano quelle polmonari e si ha un'inversione rispetto alla situazione fetale. Quindi la direzione del flusso ematico attraverso il dotto arterioso pervio si inverte, creando uno shunt ematico sn-dx (circolazione di transizione). Questa situazione persiste da subito dopo la nascita (quando aumenta il flusso ematico polmonare e si verifica la chiusura funzionale del forame ovale) fino a circa 24-72 h di vita, quando il dotto arterioso si comprime. Il sangue proveniente dall'aorta che entra nel dotto arterioso e nei suoi vasa vasorum ha un'alta PO2 che, insieme alle variazioni nel metabolismo delle prostaglandine, determina vasocostrizione e chiusura del dotto arterioso. Una volta che il dotto arterioso si chiude, si instaura la circolazione di tipo adulto. I 2 ventricoli ora pompano in serie e non esistono shunt rilevanti tra circolazione polmonare e sistemica.
Durante i primi giorni di vita, in caso di distress neonatale si può avere il ritorno a una circolazione di tipo fetale. L'asfissia con ipossia e ipercapnia causa vasocostrizione delle arteriole polmonari e vasodilatazione del dotto arterioso, con inversione dei processi descritti precedentemente e con la realizzazione di uno shunt destro-sinistro attraverso il dotto arterioso nuovamente pervio e/o il forame ovale riaperto. Di conseguenza, il neonato presenta una grave ipossiemia, condizione, questa, denominata ipertensione polmonare persistente o circolazione fetale persistente (nonostante non vi sia circolazione ombelicale). Il trattamento si propone di rimuovere le cause che causano la vasocostrizione polmonare.
Riferimento per la circolazione fetale normale
1. Koutroulou I, Tsivgoulis G, Tsalikakis D, et al: Epidemiology of Patent Foramen Ovale in General Population and in Stroke Patients: A Narrative Review. Front Neurol 11:281, 2020. Pubblicato il 28/04/2020 doi:10.3389/fneur.2020.00281
Fisiopatologia delle anomalie cardiache congenite
Le anomalie cardiache congenite sono classificate (vedi tabella Classificazione delle cardiopatie congenite del cuore) come
Cianogena
Non cianogena (shunt sinistro-destro o lesioni ostruttive)
Le conseguenze fisiologiche delle anomalie cardiache congenite variano notevolmente, da un soffio cardiaco o discrepanza dei polsi in un bambino asintomatico oppure un battito cardiaco anomalo sino a cianosi grave, scompenso cardiaco o collasso circolatorio.
Classificazione delle cardiopatie congenite del cuore*
Classificazione | Esempi |
|---|---|
Cianogena | |
— | Trasposizione delle grandi arterie |
Non cianogena | |
Shunt sinistro-destro | Difetto del setto interventricolare (comunicazione interventricolare) Difetto del setto interventricolare (comunicazione interventricolare) |
Ostruttivo | Sindrome del cuore sinistro ipoplasico (si manifesta spesso anche con cianosi, che può essere lieve) |
*In ordine decrescente di frequenza approssimativo. | |
Cardiopatie cianogene
Quantità variabili di sangue venoso deossigenato sono deviate al cuore sinistro (shunt destro-sinistro), riducendo la saturazione arteriosa sistemica di ossigeno.
Vi è presenza di cianosi in caso di concentrazione > 5 g/dL (> 50 g/L) di emoglobina deossigenata. Le complicanze di una cianosi persistente comprendono la policitemia, ippocratismo digitale, tromboembolismo (ictus incluso), disturbi emorragici, ascesso cerebrale e iperuricemia. Le crisi ipercianotiche possono verificarsi nei neonati con tetralogia di Fallot non riparata o altri difetti congeniti complessi con stenosi sottopolmonare dinamica e un difetto ventricolare.
A seconda della malformazione, il flusso ematico polmonare può essere ridotto, normale o aumentato (spesso sfociando in insufficienza cardiaca in aggiunta alla cianosi), risultando in cianosi di variabile entità. I soffi cardiaci sono variamente auscultabili e non sono specifici.
Shunt sinistro-destro
Consiste in sangue ossigenato dal cuore sinistro (atrio sinistro o ventricolo sinistro) o in uno shunt aortico al cuore destro (atrio o ventricolo destro) o dall'arteria polmonare attraverso una breccia o una comunicazione tra i 2 lati.
Subito dopo la nascita, la resistenza vascolare polmonare è elevata e il flusso attraverso questa comunicazione può essere minimo o bidirezionale. Tuttavia, entro le prime 24/48 h di vita la resistenza vascolare polmonare scende progressivamente e, a quel punto, il sangue inizierà sempre di più a scorrere da sinistra a destra. L'ulteriore afflusso di sangue al lato destro aumenta il flusso ematico polmonare e la pressione arteriosa polmonare di vario grado. Maggiore è l'aumento, più gravi sono i sintomi; un piccolo shunt sinistro-destro tipicamente non provoca sintomi o segni.
Gli shunt ad alta pressione (quelli a livello ventricolare oppure delle grandi arterie) compaiono diversi giorni o alcune settimane dopo la nascita; gli shunt a bassa pressione (difetti del setto interatriale) compaiono considerevolmente più tardi. Se non trattati, i relativi flusso ematico e pressione arteriosa polmonare elevate possono portare a malattia vascolare polmonare e finalmente alla sindrome di Eisenmenger. Grandi shunt sinistro-destro (p. es., ampio difetto del setto interventricolare (comunicazione interventricolare), dotto arterioso pervio) causano un eccesso di flusso sanguigno polmonare e sovraccarico di volume del ventricolo sinistro, che possono portare a segni di insufficienza cardiaca, che durante l'infanzia si traduce spesso in difficoltà di accrescimento. Inoltre, un ampio shunt sinistro-destro porta a una compliance polmonare inferiore e a una maggiore resistenza delle vie respiratorie. Questi fattori aumentano la probabilità di ospedalizzazione nei neonati con virus respiratorio sinciziale o altre infezioni del tratto respiratorio superiore o inferiore.
Lesioni ostruttive
Il flusso sanguigno è ostruito, causando un gradiente pressorio attraverso l'ostruzione.
Il risultante sovraccarico pressorio a monte dell'ostruzione può causare ipertrofia ventricolare e insufficienza cardiaca. La più ovvia delle manifestazioni è un soffio cardiaco, che deriva dal flusso turbolento attraverso il punto ostruito (stenotico). Esempi sono la stenosi aortica congenita, che rappresenta il 3-6% delle cardiopatie congenite e la stenosi polmonare congenita, che rappresenta l'8-12% (1, 2).
Insufficienza cardiaca
Alcune cardiopatie (p. es., valvola aortica bicuspide, stenosi aortica lieve) non alterano significativamente l'emodinamica. Altre anomalie causano sovraccarico di pressione o di volume, talvolta causando insufficienza cardiaca. L'insufficienza cardiaca si verifica quando la gittata cardiaca è insufficiente a soddisfare le esigenze metaboliche dell'organismo o quando il cuore non riesce a gestire adeguatamente il ritorno venoso, causando congestione polmonare (nell'insufficienza ventricolare sinistra), edema soprattutto nei tessuti dipendenti e nei visceri addominali (nell'insufficienza ventricolare destra), o in entrambi. L'insufficienza cardiaca nei neonati e nei bambini ha varie cause oltre alle anomalie cardiache congenite (vedi tabella Cause frequenti di insufficienza cardiaca nei bambini).
Cardiopatia congenita dotto-dipendente
Il dotto arterioso è una normale connessione tra l'arteria polmonare e l'aorta; è necessario per una corretta circolazione fetale. Alla nascita, l'aumento della PaO2 e il calo della concentrazione di prostaglandine, causa chiusura del dotto arterioso che inizia in genere entro le prime 10-15 h di vita.
Alcuni disturbi cardiaci congeniti dipendono dal dotto arterioso che rimane aperto per mantenere il flusso di sangue sistemico (p. es., sindrome del cuore sinistro ipoplasico, stenosi aortica critica, coartazione aortica) che per il flusso di sangue polmonare (lesioni cianotiche come atresia polmonare o grave tetralogia di Fallot). Mantenere il dotto arterioso pervio attraverso l'infusione di prostaglandine è quindi di vitale importanza in questi disturbi prima della riparazione definitiva (di solito chirurgica).
Riferimenti relativi alla fisiopatologia
1. Daubeney PEF, Rigby ML, Niwa K, Gatzoulis MA (eds): Pediatric Heart Disease: A Practical Guide. Wiley-Blackwell 2012.
2. Hoffman JI, Kaplan S: The incidence of congenital heart disease. J Am Coll Cardiol 39(12):1890-1900, 2002. doi:10.1016/s0735-1097(02)01886-7
Sintomatologia delle malattie cardiache congenite
Le manifestazioni delle cardiopatie congenite sono varie ma comunemente includono
Soffi
Cianosi
Insufficienza cardiaca
Impulsi diminuiti o non palpabili
Altre anomalie presenti all'esame obiettivo possono includere shock circolatorio, scarsa perfusione, secondo tono cardiaco anormale (S2, singolo o ampiamente separato), click sistolico, galoppo, o ritmo lento in maniera anomala, veloce, o irregolare.
Soffi
La maggior parte degli shunt sinistro-destro e delle lesioni ostruttive causa soffi sistolici. I soffi e i fremiti cardiaci sono meglio apprezzabili in corrispondenza della superficie più vicina al loro punto di origine e attraverso la loro localizzazione sono di grande aiuto nella diagnosi. Un aumento del flusso attraverso la valvola polmonare o aortica provoca un soffio crescendo-decrescendo mesosistolico (eiezione sistolica). Un flusso da insufficienza attraverso una valvola atrioventricolare o il flusso all'interno di un difetto del setto interventricolare provoca un soffio olosistolico (pansistolico) che nasconde il primo tono cardiaco (S1) all'aumento della sua intensità.
Il dotto arterioso pervio in genere provoca un soffio continuo che non è interrotto dalL'S2 perché il sangue scorre attraverso il dotto durante la sistole e la diastole. Questo soffio ha un 2o tono, avente un suono più pronunciato durante la sistole rispetto alla diastole (quando è spinto da una pressione maggiore).
Cianosi

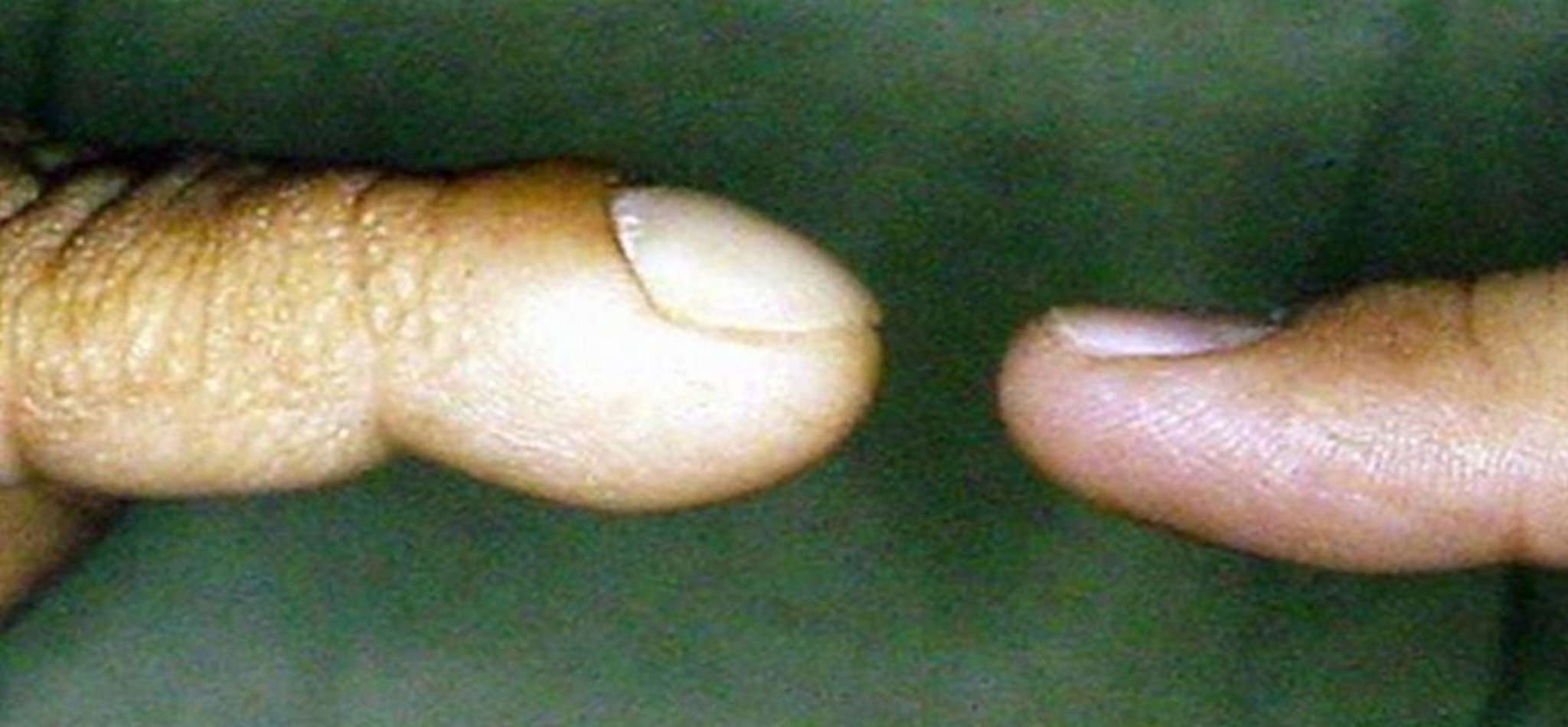
© Springer Science+Business Media
La cianosi centrale è caratterizzata da un colore bluastro delle labbra e della lingua e/o del letto ungueale; si verifica quando c'è un aumento dell'emoglobina deossigenata (almeno 5 g/dL [50 g/L]) e implica un basso livello del contenuto ematico di ossigeno (di solito saturazione di ossigeno < 85%). La cianosi periorale e acrocianosi (cianosi delle mani e dei piedi) senza cianosi delle labbra o del letto ungueale è causata da vasocostrizione periferica piuttosto che ipossiemia ed è un comune reperto normale nei neonati. I bambini più grandi con cianosi di vecchia data sviluppano spesso ippocratismo digitale.
Insufficienza cardiaca
Nei neonati i sintomi o segni di insufficienza cardiaca comprendono
Tachicardia
Tachipnea
Dispnea durante l'alimentazione
Diaforesi, in particolare durante l'alimentazione
Irrequietezza, irritabilità
Epatomegalia
La dispnea durante l'alimentazione causa un apporto inadeguato di nutrienti e scarsa crescita, che può essere peggiorata per maggiori richieste metaboliche nell'insufficienza cardiaca e frequenti infezioni delle vie respiratorie. Al contrario degli adulti e dei bambini più grandi, la maggior parte dei neonati non presenta distensione delle vene del collo né edemi declivi; tuttavia talvolta presentano edema nell'area periorbitale. L'epatomegalia è una caratteristica particolarmente importante dell'insufficienza cardiaca nei neonati a causa della distensibilità della capsula epatica a questa età. I segni presenti nei bambini più grandi con insufficienza cardiaca sono simili a quelli degli adulti.
Altre manifestazioni dei difetti cardiaci
Nei neonati, lo shock circolatorio può essere il primo sintomo di alcune anomalie (p. es., sindrome del cuore sinistro ipoplasico, stenosi aortica critica, arco aortico interrotto, coartazione dell'aorta). I neonati appaiono molto malati con mucose pallide o cianotiche, estremità fredde, scomparsa del battito, pressione arteriosa bassa e ridotta risposta agli stimoli.
Il dolore toracico nei bambini di solito non è cardiaco. Nei neonati, il dolore toracico può manifestarsi con inspiegabile marcata irritabilità, soprattutto durante o dopo l'allattamento, e può essere causato da origine anomala dell'arteria coronaria sinistra dall'arteria polmonare. Nei bambini più grandi e negli adolescenti, il dolore toracico dovuto a un'eziologia cardiaca è in genere associato allo sforzo e può essere causato da un'anomalia coronarica, da pericardite, miocardite, cardiomiopatia ipertrofica o stenosi aortica severa.
La sincope, tipicamente senza pericolosi sintomi e spesso in associazione con lo sforzo, si può verificare con alcune anomalie tra cui una cardiomiopatia (ipertrofica o dilatata), l'origine anomala delle coronarie o sindromi aritmiche ereditarie (p. es., sindrome del QT lungo, tachicardia ventricolare polimorfica catecolaminergica [CPVT], sindrome di Brugada). Gli atleti in età scolare sono colpiti più comunemente.
Diagnosi delle malattie cardiache congenite
Controllo con pulsossimetria
Esame cardiaco obiettivo
RX torace ed ECG
Ecocardiografia
A volte RM cardiaca o angio-TC, cateterismo cardiaco con angiocardiografia
Quando presenti, soffi cardiaci, cianosi, polsi anormali o manifestazioni di insufficienza cardiaca suggeriscono cardiopatie congenite. Nei neonati con questi segni, si esegue l'ecocardiografia che conferma la diagnosi di cardiopatie congenite. Se l'unica anomalia è la cianosi, deve essere anche esclusa la presenza di metemoglobinemia.
Sebbene l'ecocardiografia sia tipicamente diagnostica, in alcuni casi, la RM cardiaca o l'angio-TC possono chiarire importanti dettagli anatomici. Il cateterismo cardiaco con l'angiocardiografia è solitamente necessario per confermare la diagnosi o per valutare la gravità dell'anomalia; tuttavia, è generalmente effettuato solo per scopi terapeutici.
Screening neonatale per cardiopatie congenite
Nei neonati le manifestazioni di cardiopatie congenite possono essere discrete o assenti e la mancata o ritardata rilevazione di una cardiopatia congenita, può portare a mortalità neonatale o significativa morbilità, in particolare nel 10-15% dei neonati che necessitano di un trattamento chirurgico o di un trattamento medico in regime di ricovero nelle prime ore o nei primi giorni di vita. Pertanto, lo screening universale per le malattie cardiache congenite critiche con pulsossimetria è raccomandato per tutti i neonati prima della dimissione ospedaliera (1). Lo screening è eseguito nei neonati con ≥ 24 h di vita ed è considerato positivo se ≥ 1 dei seguenti segni è presente:
Qualsiasi misurazione della saturazione di ossigeno è < 90%.
Le misure della saturazione di ossigeno nella mano e nel piede destri sono < 95% su 3 misurazioni separate ottenute ad 1 h di distanza.
Vi è > 3% di differenza assoluta tra la saturazione di ossigeno della mano destra (preduttale) e del piede (postduttale) su 3 misurazioni abbinate, separate prese a 1 h di distanza.
Tutti i neonati con un risultato di screening positivo devono essere sottoposti a una valutazione completa per cardiopatie congenite e altre cause di ipossiemia (p. es., vari disturbi respiratori, depressione del sistema nervoso centrale, sepsi) comprendendo tipicamente una RX torace, ECG, ecocardiogramma e spesso l'esame del sangue. La sensibilità dello screening con pulsossimetria è leggermente > 75%; le più frequenti mancate diagnosi di lesioni di cardiopatie congenite sono le lesioni ostruttive del cuore sinistro (p. es., coartazione aortica).
Riferimento per lo screening neonatale
1. Martin GR, Ewer AK, Gaviglio A, et al: Updated Strategies for Pulse Oximetry Screening for Critical Congenital Heart Disease. Pediatrics 146(1):e20191650, 2020. doi:10.1542/peds.2019-1650
Trattamento delle malattie cardiache congenite
Stabilizzazione medica dell'insufficienza cardiaca (p. es., con diuretici, ACE-inibitori, beta-bloccanti, digossina, spironolattone, restrizione salina e, in casi selezionati, ossigeno supplementare o prostaglandina E1)
Riparazione chirurgica o intervento transcatetere
Il trattamento dell'insufficienza cardiaca varia ampiamente a seconda dell'eziologia. La terapia definitiva generalmente richiede la correzione del problema di base.
Dopo la stabilizzazione medica dei sintomi acuti di insufficienza cardiaca o della cianosi, la maggior parte dei bambini necessita di riparazione chirurgica o transcatetere; le eccezioni sono alcuni difetti del setto interventricolare (comunicazione interventricolare) che potrebbero diventare più piccoli, chiudersi con il tempo oppure evolvere in una disfunzione valvolare lieve. Le procedure transcatetere comprendono
Settostomia atriale con pallone per palliazione di neonati gravemente cianotici con trasposizione delle grandi arterie
Dilatazione con palloncino della stenosi della valvola aortica grave o della stenosi della valvola polmonare
Chiusura transcatetere di shunt cardiaci (il più delle volte difetto del setto interatriale e del dotto arterioso pervio)
Posizionamento transcatetere della valvola polmonare
Dilatazione con palloncino con o senza stenting delle stenosi vascolari, più comunemente le stenosi dell'arteria polmonare
Insufficienza cardiaca nei neonati
L'insufficienza cardiaca acuta, grave o la cianosi sono un emergenza medica durante la prima settimana di vita. Si deve reperire un accesso vascolare, preferibilmente tramite un catetere venoso ombelicale.
Quando è sospettata o confermata una cardiopatia congenita critica, deve essere avviata un'infusione EV di prostaglandina E1 da 0,05 a 0,1 mcg/kg/min. Mantenere aperto il dotto è importante perché la maggior parte delle lesioni cardiache che si manifestano a questa età è dotto-dipendente sia per il flusso di sangue sistemico (p. es., la sindrome del cuore sinistro ipoplasico, la stenosi aortica critica, la coartazione aortica) che per il flusso di sangue polmonare (lesioni cianotiche come la stenosi polmonare critica, l'atresia polmonare o una grave tetralogia di Fallot).
La ventilazione meccanica è spesso necessaria nei neonati malati critici. L'ossigeno supplementare deve essere somministrato giudiziosamente o addirittura negato perché l'ossigeno supplementare può diminuire la resistenza vascolare polmonare, la quale è dannosa per i neonati con alcuni difetti (p. es., sindrome del cuore sinistro ipoplasico).
Altre terapie per l'insufficienza cardiaca neonatale includono diuretici, farmaci inotropi e farmaci per ridurre il postcarico. Il diuretico furosemide viene somministrato con un bolo iniziale di 1 mg/kg EV e titolato in base alla diuresi. L'infusione di inotropi come dopamina o dobutamina è in grado di supportare la pressione arteriosa ma ha lo svantaggio di aumentare la frequenza cardiaca e il postcarico, aumentando così il consumo di ossigeno del miocardio. Sono utilizzati raramente in neonati con cardiopatia congenita. Il milrinone è sia un inotropo positivo che un vasodilatatore ed è spesso utilizzato nel postoperatorio nei pazienti con cardiopatie congenite. La dopamina, la dobutamina e milrinone hanno il potenziale di aumentare il rischio di aritmie. Il nitroprussiato, un vasodilatatore puro, può essere utilizzato per l'ipertensione postoperatoria. È infuso inizialmente a 0,3-0,5 mcg/kg/min e titolato all'effetto desiderato (la dose di mantenimento usuale è di circa 3 mcg/kg/min).
Insufficienza cardiaca nei lattanti più grandi e bambini
Le terapie spesso comprendono un diuretico (p. es., furosemide 0,5-1 mg/kg EV o da 1 a 3 mg/kg per via orale ogni 8-24 h, aumentando il dosaggio se necessario), e un ACE-inibitore (p. es., captopril da 0,1 a 0,3 mg/kg per via orale 3 volte/die). Può essere utile un diuretico risparmiatore di potassio (p. es., spironolattone 1 mg/kg per via orale 1 o 2 volte/die, titolato fino a 2 mg/kg/dose, se necessario), soprattutto se sono richieste alte dosi di furosemide. I beta-bloccanti (p. es., carvedilolo, metoprololo) sono spesso aggiunti per i bambini con insufficienza cardiaca congestizia cronica. I nuovi farmaci utilizzati negli adulti per l'insufficienza cardiaca, come il sacubitril/valsartan e gli inibitori del sodium-glucose cotransporter-2 (SGLT-2), possono essere utili, ma i dati sono limitati nella popolazione pediatrica (1).
La digossina è usata meno spesso che in passato, ma può ancora avere un ruolo nei bambini con insufficienza cardiaca che hanno grandi shunt sinistra/destra e in alcuni pazienti con cardiopatie congenite nel postoperatorio (la dose varia a seconda dell'età; vedi tabella Posologia di digossina orale nei bambini). In particolare, è stato dimostrato che la digossina riduce la mortalità nei pazienti con ventricolo singolo dopo la procedura di Norwood e prima dell'intervento chirurgico di seconda fase (2). L'uso della digossina come farmaco di prima linea nel trattamento della tachicardia sopraventricolare neonatale è diminuito perché provoca una mortalità più elevata rispetto al trattamento con propranololo (3). Tuttavia, se la sindrome di Wolff-Parkinson-White non è presente, può essere utile come agente primario se il propranololo è inefficace o come secondo agente combinato con il propranololo o con altri farmaci antiaritmici.
L'ossigeno supplementare può diminuire l'ipossiemia e alleviare il distress respiratorio nell'insufficienza cardiaca; quando possibile, la frazione di inspirazione di ossigeno (FiO2) deve essere mantenuta < 40% per minimizzare il rischio di danno epiteliale polmonare. L'ossigeno supplementare deve essere usato con cautela, se impiegato, in pazienti con lesioni da shunt sinistro-destro o malattia ostruttiva cardiaca sinistra perché può esacerbare il sovraccarico del circolo polmonare.
In generale, è consigliabile una dieta sana, compresa la restrizione di sale, anche se possono essere necessarie modifiche dietetiche a seconda del disturbo e delle manifestazioni specifiche. L'insufficienza cardiaca aumenta le richieste metaboliche e la corrispondente dispnea rende l'alimentazione più difficile. Nei neonati con malattie cardiache congenite critiche, in particolare quelli con lesioni ostruttive del cuore sinistro, l'alimentazione può essere sospesa per ridurre al minimo il rischio di enterocolite necrotizzante. Nei neonati con insufficienza cardiaca dovuta a shunt sinistro-destro, è raccomandato l'aumento del contenuto calorico delle poppate; questo tipo di nutrizione aumenta le calorie fornite, con meno rischi di sovraccarico di volume. Alcuni bambini richiedono alimentazione enterale per mantenere lo sviluppo. Se con questi accorgimenti non si ottiene l'aumento di peso, è indicata la riparazione chirurgica dell'anomalia.
Profilassi dell'endocardite
Le linee guida dell'American Heart Association per la prevenzione dell'endocardite (4) affermano che la profilassi antibiotica è necessaria per i bambini con cardiopatie congenite che hanno i seguenti:
Cardiopatie congenite cianogene non riparate (compresi i bambini con shunt e condotti palliativi)
Cardiopatie congenite completamente riparate dopo l'intervento chirurgico durante i primi 6 mesi se è stato utilizzato materiale protesico o un dispositivo
Cardiopatia congenita riparata con difetti residui sul sito o adiacenti a un patch chirurgico o un dispositivo protesico
Valvola meccanica o bioprotetica
Precedente episodio di endocardite
Riferimenti relativi al trattamento
1. Loss KL, Shaddy RE, Kantor PF: Recent and Upcoming Drug Therapies for Pediatric Heart Failure. Front Pediatr 9:681224, 2021. Pubblicato l'1/11/ 2021 doi:10.3389/fped.2021.681224
2. Oster ME, Kelleman M, McCracken C, et al: Association of digoxin with interstage mortality: Results from the Pediatric Heart Network Single Ventricle Reconstruction Trial Public Use Dataset. J Am Heart Assoc 5(1): e002566., 2016.
3. Bolin EH, Lang SM, Tang X, et al: Propranolol versus digoxin in the neonate for supraventricular tachycardia (from the Pediatric Health Information System). Am J Cardiol 119(10): 1605–1610, 2017.
4. Baltimore RS, Gewitz M, Baddour LM, et al: Infective Endocarditis in Childhood: 2015 Update: A Scientific Statement From the American Heart Association. Circulation 132(15):1487–1515, 2015. doi:10.1161/CIR.0000000000000298
Per ulteriori informazioni
Le seguenti risorse in lingua inglese possono essere utili. Si noti che il Manuale non è responsabile per il contenuto di queste risorse.
American Heart Association: Common Heart Defects: fornisce una panoramica dei difetti cardiaci congeniti comuni per genitori e tutori
American Heart Association: Infective Endocarditis: fornisce una panoramica dell'endocardite infettiva, inclusa una sintesi dell'uso profilattico di antibiotici, per pazienti e tutori







