


L'amiloidosi è una condizione di un gruppo disparato di patologie caratterizzate dalla deposizione extracellulare di fibrille insolubili composte da proteine disaggregate. Queste proteine si possono accumulare localmente, causando relativamente pochi sintomi, oppure in modo diffuso, coinvolgendo molti organi e causando una grave insufficienza multiorgano. L'amiloidosi può verificarsi de novo o essere secondaria a varie infezioni, condizioni infiammatorie o neoplastiche. La diagnosi è basata sulla biopsia del tessuto colpito; la proteina amiloidogenica viene tipizzata utilizzando una varietà di tecniche immunoistologiche e biochimiche. La terapia cambia in base al tipo di amiloidosi.
Le fibrille di amiloide sono composte da proteine normalmente solubili mal ripiegate che si aggregano in oligomeri e poi fibrille insolubili. Un certo numero di proteine normali (wild-type) e mutanti sono suscettibili a tale ripiegamento e aggregazione (proteine amiloidogeniche), così da giustificare la grande varietà di cause e tipi di amiloidosi.
I depositi di amiloide sono composti da piccole fibrille insolubili (circa 10 nm di diametro) che formano foglietti congofili beta-plissettati che possono essere identificati mediante diffrazione dei raggi X. Oltre alla proteina fibrillare amiloide, i depositi contengono anche un componente dell'amiloide P sierica e glicosaminoglicani.
I depositi di amiloide si colorano di rosa con l'ematossilina eosina e contengono componenti di carboidrati che si colorano con l'acido periodico di Schiff o con l'Alcian blu, ma la maggior parte tipicamente ha birifrangenza color mela verde al microscopio a luce polarizzata dopo colorazione col rosso Congo. All'osservazione autoptica, gli organi colpiti possono apparire cerei.
Per sviluppare amiloidosi, oltre alla produzione di proteine amiloidogeniche, probabilmente c'è anche un fallimento dei normali meccanismi di eliminazione di tali proteine mal ripiegate. Gli stessi depositi di amiloide sono metabolicamente inerti ma interferiscono fisicamente con la struttura e la funzione degli organi. Tuttavia, alcuni oligomeri prefibrillari di proteine amiloidogeniche hanno tossicità cellulare diretta, una componente importante della patogenesi della malattia.
Eziologia dell'amiloidosi
Nell'amiloidosi sistemica, proteine circolanti amiloidogeniche formano depositi in diversi organi. I tipi sistemici principali comprendono
Amiloidosi primaria: causata da iperespressione acquisita di catene leggere delle immunoglobuline clonali
Amiloidosi familiare: causata dall'eredità di un gene mutante che codifica per una proteina incline al mal ripiegamento, più comunemente la transtiretina (TTR)
Amiloidosi da transtiretina wild-type (ATTRwt) (precedentemente denominata "amiloidosi sistemica senile" o SSA): causata da un mal ripiegamento e dall'aggregazione della transtiretina (TTR) "wild-type"
Amiloidosi secondaria: causata dall'aggregazione di una proteina della fase acuta, amiloide sierica A
Amiloidosi causata dall'aggregazione di beta2-microglobulina può verificarsi nei pazienti in emodialisi a lungo termine, ma l'incidenza è diminuita con l'uso delle moderne membrane da dialisi ad alto flusso. Esiste una rara forma ereditaria di amiloidosi da beta-2-microglobulina dovuta ad una mutazione del gene interessato.
Forme localizzate di amiloidosi sembrano essere causate dalla produzione locale e deposizione di una proteina amiloidogenica (in genere catene leggere delle immunoglobuline) all'interno dell'organo interessato piuttosto che dalla deposizione di proteine circolanti. Siti coinvolti sono, frequentemente, il sistema nervoso centrale (p. es., nella malattia di Alzheimer), la cute, le vie respiratorie superiori o inferiori, il parenchima polmonare, la vescica, gli occhi e le mammelle.
Amiloidosi primaria
L'amiloidosi primaria è causata da iperproduzione di una catena leggera amiloidogenica delle immunoglobuline nei pazienti con gammeopatie monoclonali o altre malattie linfoproliferative delle cellule B. Catene leggere possono anche formare depositi tissutali nonfibrillari (ossia, malattia da deposito di catene leggere). Raramente, le catene pesanti delle immunoglobuline formano fibrille amiloidi (chiamata amiloidosi AH).
Sedi frequenti di deposito di amiloide comprendono la cute, i nervi, il cuore, il tratto gastrointestinale (inclusa la lingua), i reni, il fegato, la milza e i vasi sanguigni. Di solito, una plasmocitosi di basso grado è presente nel midollo osseo, che è simile a quella di un mieloma multiplo, anche se la maggior parte dei pazienti non presenta un vero e proprio mieloma multiplo (con lesioni litiche a carico delle ossa, ipercalcemia, depositi tubulari renali, e anemia). Tuttavia, circa il 10-20% dei pazienti con mieloma multiplo sviluppa amiloidosi primaria.
Amiloidosi familiare
L'amiloidosi familiare è causata dall'ereditarietà di un gene che codifica per una proteina sierica incline all'aggregazione mutata, di solito una proteina abbondantemente prodotta dal fegato.
Proteine sieriche che possono causare amiloidosi familiare sono: la trantiretina, l'apolipoproteina A-I, l'apolipoproteina A-II, il lisozima, il fibrinogeno, la gelsolina e la cistatina C. Una forma che si ipotizza essere familiare, è causata dalla proteina del siero fattore chemiotattico dei leucociti 2 (leukocyte chemotactic factor 2 [LECT2]); tuttavia, una specifica mutazione genetica ereditaria per quest'ultimo tipo non è stata chiaramente dimostrata.
L'amiloidosi da transtiretina (ATTR) è il tipo più comune di amiloidosi familiare. Sono state associate ad amiloidosi più di 130 mutazioni del gene TTR. La mutazione più diffusa, V30M, è comune in Portogallo, Svezia, Brasile e Giappone, e una mutazione V122I è presente in circa il 4% dei neri americani e caraibici. La penetranza della malattia e l'età di insorgenza sono molto variabili, ma sono simili all'interno delle famiglie e dei gruppi etnici (1).
L'amiloidosi da transtiretina causa neuropatia periferica sensitivomotoria e neuropatia autonomica, malattia renale cronica e cardiomiopatia. La sindrome del tunnel carpale precede di solito altre manifestazioni della malattia neurologica. I depositi vitreali possono svilupparsi a causa della produzione di transitretina mutante da parte dell'epitelio retinico, oppure si possono sviluppare depositi leptomeningei come il plesso coroideo produce transitretina mutante. Quando la cardiomiopatia è la manifestazione predominante della deposizione di transtiretina (TTR) nel cuore, si parla di cardiomiopatia amiloide da transtiretina (ATTR-CM).
Amiloidosi da transtiretina wild type (ATTRwt) (amiloidosi sistemica senile)
L'amiloidosi da transtiretina wild-type (ATTRwt) è causata dall'aggregazione e dalla deposizione di transtiretina wild-type, soprattutto nel cuore.
L'amiloidosi da transtiretina wild type (ATTRwt) è sempre più riconosciuta come causa di cardiomiopatia infiltrativa negli uomini anziani. Circa il 16% dei pazienti con stenosi aortica sottoposti a sostituzione transcatetere della valvola aortica (2) e il 13% di quelli ospedalizzati per insufficienza cardiaca con frazione di eiezione conservata hanno anche una cardiomiopatia amiloide da transtiretina, in questo caso designata come wATTR-CM per indicare il deposito di transtiretina (TTR) wild-type nel cuore (3). Le manifestazioni nei tessuti molli dell'amiloidosi da transtiretina wild type (ATTRwt), tra cui la sindrome del tunnel carpale, la rottura del tendine bicipitale, le rotture della cuffia dei rotatori e la stenosi spinale, possono precedere di anni l'espressione clinica della cardiomiopatia infiltrativa.
I fattori genetici ed epigenetici che portano all'amiloidosi da transtiretina wild type (ATTRwt) sono sconosciuti. Poiché l'amiloidosi da transtiretina wild type (ATTRwt) e amiloidosi primaria possono causare cardiomiopatia, e che gammopatie monoclonali possono essere presenti nei pazienti in questa fascia di età, è indispensabile individuare con precisione il tipo di amiloide in modo che i pazienti con amiloidosi da transtiretina wild type non siano impropriamente trattati con chemioterapia (che è utilizzata per l'amiloidosi primaria).
Amiloidosi AA (amiloidosi secondaria)
Questa forma può insorgere secondariamente a varie patologie infettive, infiammatorie e neoplastiche ed è causata dall'aggregazione di isoforme della proteina della fase acuta, amiloide sierica A.
Comuni infezioni responsabili sono
Le condizioni infiammatorie predisponenti comprendono
Sindromi da febbre periodiche ereditate come febbre mediterranea familiare
Malattia di Castleman
Le citokine infiammatorie (p. es., interleuchina-1, TNF, IL-6), che sono prodotte in queste patologie o ectopicalmente tramite cellule tumorali provocano aumentata sintesi epatica dell'amiloide A sierica.
L'amiloidosi secondaria mostra una predilezione per i reni, la milza, il fegato, le ghiandole surrenali e i linfonodi. Il coinvolgimento del cuore o dei nervi periferici e autonomici si verifica in ritardo nel decorso della malattia.
Amiloidosi localizzata
L'amiloidosi localizzata al di fuori del cervello è più frequentemente causata da depositi di catene leggere immunoglobuline clonali; nel cervello la proteina beta amiloide predomina.
I depositi localizzati di amiloide tipicamente coinvolgono le vie respiratorie e i polmoni, la vescica e gli ureteri, la cute, le mammelle e gli occhi. Raramente, altre proteine prodotte localmente causano amiloidosi, come isoforme della cheratina che possono formare depositi a livello cutaneo. Le catene leggere clonali delle immunoglobuline prodotte dal tessuto linfoide associato alle mucose nel tratto gastrointestinale, nelle vie respiratorie e nella vescica possono portare a amiloidosi primaria localizzata in tali organi.
I depositi della proteina beta amiloide nel cervello contribuiscono alla malattia di Alzheimer o all'angiopatia amiloide cerebrovascolare. Altre proteine prodotte nel sistema nervoso centrale possono malripiegarsi, aggregarsi, e danneggiare neuroni provocando malattie neurodegenerative (p. es., morbo di Parkinson, malattia di Huntington).
Riferimenti relativi all'eziologia
1. Buxbaum JN, Ruberg FL: Transthyretin V122I (pV142I)* cardiac amyloidosis: an age-dependent autosomal dominant cardiomyopathy too common to be overlooked as a cause of significant heart disease in elderly African Americans. Genet Med 19(7):733-742, 2017. doi:10.1038/gim.2016.200
2. Fabbri G, Serenelli M, Cantone A, et al: Transthyretin amyloidosis in aortic stenosis: clinical and therapeutic implications. Eur Heart J Suppl 23(Suppl E):E128-E132, 2021. doi:10.1093/eurheartj/suab107
3. Magdi M, Mostafa MR, Abusnina W, et al: A systematic review and meta-analysis of the prevalence of transthyretin amyloidosis in heart failure with preserved ejection fraction. Am J Cardiovasc Dis 12(3):102-111, 2022. PMID: 35873185
Sintomatologia dell'amiloidosi
I sintomi e i segni di amiloidosi sistemica sono aspecifici, spesso con conseguente ritardo nella diagnosi. Il sospetto di amiloidosi deve aumentare nei pazienti con un progressivo processo di malattia multisistemica.
Depositi di amiloide renali si verificano in genere nella membrana glomerulare comportando proteinuria, ma in circa il 15% dei casi i tubuli sono colpiti, provocando azotemia con proteinuria soltanto minima. Questi processi possono progredire fino alla sindrome nefrotica con marcata ipoalbuminemia, edema, e anasarca ed a insufficienza renale terminale.
L'interessamento epatico causa epatomegalia indolore, che può essere di grado importante. Test di funzionalità epatica di solito suggeriscono colestasi intraepatica con elevazione della fosfatasi alcalina e più tardi della bilirubina, anche se l'ittero è raro. Occasionalmente, si ha ipertensione portale e, di conseguenza, varici esofagee e ascite.
Il coinvolgimento delle vie aeree e laringee porta a dispnea, raucedine, respiro affannoso, emottisi, o ostruzione delle vie aeree.
L'infiltrazione del miocardio provoca una cardiomiopatia restrittiva, portando infine alla disfunzione diastolica ed all'insufficienza cardiaca; possono verificarsi blocco cardiaco o aritmia. L'ipotensione è frequente.
La neuropatia periferica, con parestesie delle dita dei piedi e delle mani, è un sintomo frequente d'esordio nelle amiloidosi primaria e amiloidosi da transtiretina (ATTR). La neuropatia autonomica può causare ipotensione ortostatica, disfunzione erettile, alterazione della sudorazione, ritenzione urinaria e disturbi della motilità gastrointestinale.
L'angiopatia cerebrovascolare amiloide è il più delle volte causa di emorragia cerebrale spontanea ma alcuni pazienti hanno brevi, sintomi neurologici transitori.
L'amiloidosi gastrointestinale può causare anomalie della motilità dell'esofago e del piccolo e grande intestino. Può anche verificarsi atonia gastrica, malassorbimento, sanguinamento o una pseudo-ostruzione. La macroglossia è frequente nell'amiloidosi primaria.
Il coinvolgimento dei tessuti molli amiloidi è caratteristicamente precedente all'espressione clinica della cardiomiopatia amiloide da amiloidosi da transtiretina wild type (ATTRwt). Le manifestazioni della malattia amiloide dei tessuti molli comprendono la sindrome del tunnel carpale, il dito a scatto, la rottura del tendine bicipitale e la stenosi spinale.

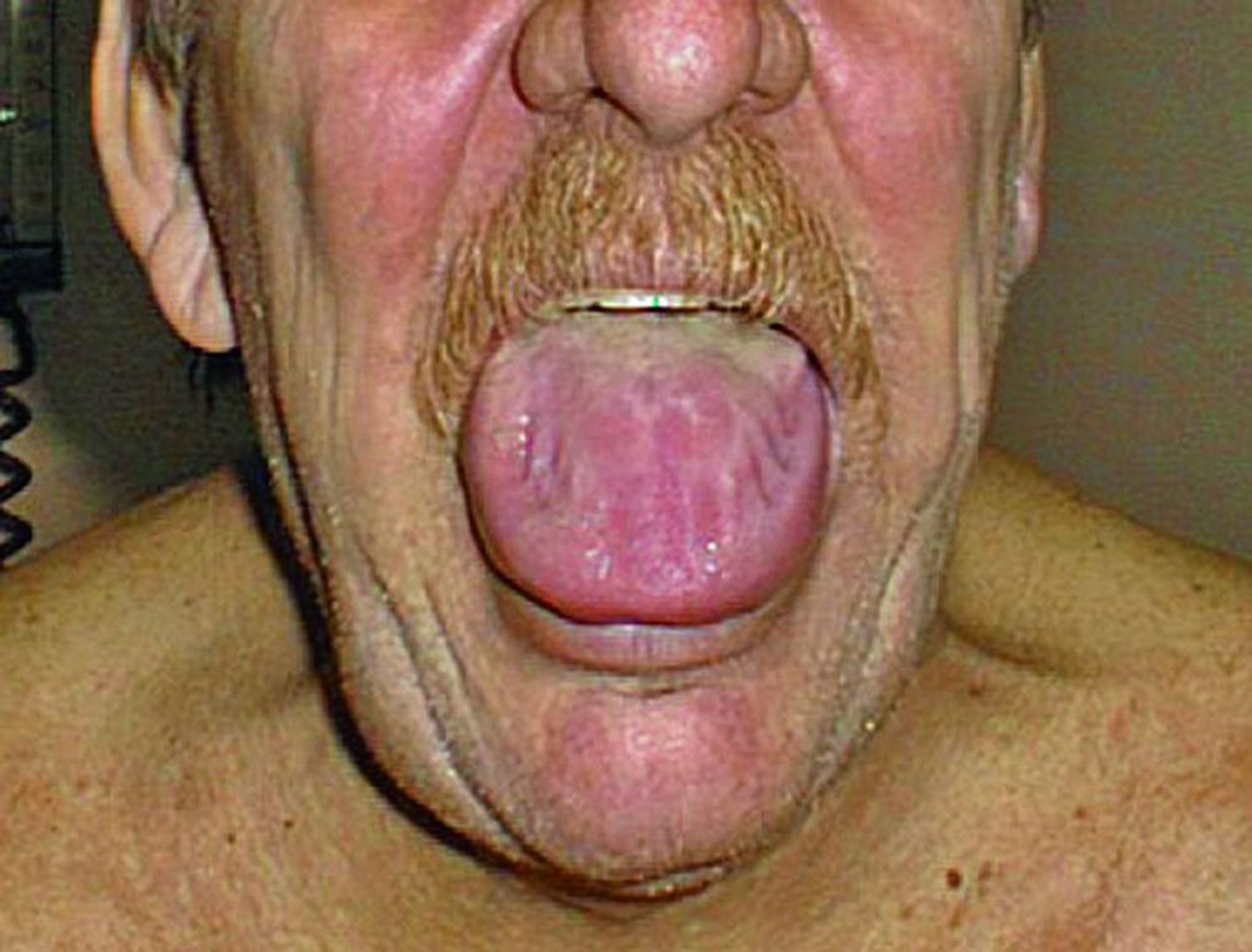
© Springer Science+Business Media
L'amiloidosi della tiroide può causare un gozzo fermo, simmetrico, non dolente simile a quello trovato nella tiroidite di Hashimoto. Possono anche verificarsi altre endocrinopatie.
L'interessamento polmonare (soprattutto nell'amiloidosi primaria) può essere caratterizzato da noduli e cisti polmonari focali, lesioni tracheobronchiali, versamenti pleurici o da depositi alveolari-settali (interstiziali) diffusi.
L'opacità amiloide vitreale e i margini pupillari bilaterali smerlati si sviluppano in numerose amiloidosi ereditarie.
Altre manifestazioni includono lividi, spesso intorno agli occhi (occhi di procione), causati da depositi di amiloide nei vasi sanguigni. I depositi di amiloide causano l'indebolimento dei vasi sanguigni, che possono rompersi dopo un trauma minore, come starnuti o tosse.
Diagnosi dell'amiloidosi
Biopsia
Tipizzazione amiloide
Test per il coinvolgimento degli organi
Biopsia
La diagnosi di amiloidosi è basata sulla dimostrazione di depositi fibrillari in un organo coinvolto. L'aspirazione di grasso addominale sottocutaneo rileva i depositi di amiloide in circa l'80% dei pazienti con amiloidosi primaria ma in meno del 25% nei pazienti con amiloidosi transtiretina wild type (ATTRwt) (1). Se il risultato della biopsia del tessuto adiposo è negativo allora si dovrà bioptizzare l'organo clinicamente coinvolto. La sensibilità diagnostica delle biopsie renali e cardiache è quasi del 100% quando questi organi sono clinicamente coinvolti. Le sezioni di tessuto vengono colorate con rosso Congo e osservate con un microscopio a luce polarizzata per ricercare la caratteristica birifrangenza. Fibrille nonbranching di 10 nm possono anche essere riconosciute mediante microscopia elettronica su campioni bioptici di cuore o di rene.
La scintigrafia utilizzando traccianti che si attaccano all'osso può diagnosticare una cardiomiopatia da amiloide da transtiretina (ATTR) senza biopsia cardiaca, a condizione che sia esclusa l'amiloidosi primaria.
Tipizzazione amiloide
Dopo che l'amiloidosi è stata confermata dalla biopsia, il tipo è determinato utilizzando una varietà di tecniche. Per alcuni tipi di amiloidosi, l'immunoistochimica o l'immunofluorescenza possono essere diagnostiche, ma possono verificarsi anche falsi positivi. Altre tecniche utili includono il sequenziamento del gene per l'amiloidosi familiare, e l'identificazione biochimica precisa mediante spettrometria di massa delle varianti proteiche nei depositi di amiloide (il metodo più sensibile e specifico).
Se si sospetta amiloidosi primaria, i pazienti devono essere valutati per una malattia delle plasmacellule sottostante usando misurazione quantitativa nel siero delle catene leggere delle immunoglobuline, e rilevazione qualitativa nel siero o nelle urine di catene leggere monoclonali utilizzando immunofissazione (elettroforesi delle proteine nel siero e nelle urine non sono test sensibili nei pazienti con amiloidosi primaria), e una biopsia del midollo osseo con la citometria a flusso o immunoistochimica per stabilire clonalità delle plasmacellule.
I pazienti con plasmacellule clonali > 10% devono essere testati per verificare se soddisfano i criteri per il mieloma multiplo, incluso screening per lesioni ossee litiche, anemia, insufficienza renale, e ipercalcemia.
Coinvolgimento degli organi
I pazienti sono sottoposti a screening per il coinvolgimento degli organi che inizia con test non invasivi:
Reni: esame delle urine; misurazione dell'azotemia, della creatinina e dell'albumina; stima della velocità di filtrazione glomerulare; e raccolta delle urine nelle 24 h per elettroforesi delle proteine
Fegato: test di funzionalità epatica
Polmoni: RX torace, TC del torace e test di funzionalità polmonare
Cuore: elettrocardiogramma e misura dei biomarcatori come il peptide natriuretico cerebrale o del N-terminal-pro-BNP (NT-proBNP) e della troponina
Si può sospettare il coinvolgimento cardiaco dal basso voltaggio all'ECG (causato da un ventricolo infiltrato), e/o da aritmie. Se si sospetta un coinvolgimento cardiaco a causa dei sintomi, oltre ai risultati dell'ECG e ai biomarcatori cardiaci, si esegue un'ecocardiografia per misurare il rilassamento diastolico e la deformazione longitudinale del muscolo cardiaco (una misura della funzione sistolica ventricolare sinistra) e per lo screening dell'ipertrofia biventricolare. Nei casi ambigui, la RM cardiaca può rilevare un enhancement subendocardico persistente del gadolinio, un reperto caratteristico. Le scansioni nucleari cardiache con tecnezio pirofosfato migliorano il rilevamento della malattia cardiaca amiloide da accumulo di transtiretrina (ATTR) possono evitare la necessità di biopsie cardiache a condizione che gli esami del sangue escludano l'amiloidosi da catene leggere (2, 3).
Riferimenti relativi alla diagnosi
1. Aimo A, Emdin M, Musetti V, et al: Abdominal Fat Biopsy for the Diagnosis of Cardiac Amyloidosis. JACC Case Rep 2(8):1182-1185, 2020. doi:10.1016/j.jaccas.2020.05.062
2. Gillmore JD, Maurer MS, Falk RH, et al: Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation 133(24):2404–2412, 2016.
3. Maurer MS, Bokhari S, Damy T, et al: Expert consensus recommendations for the suspicion and diagnosis of transthyretin cardiac amyloidosis. Circ Heart Fail 12(9):e006075, 2019.
Trattamento dell'amiloidosi
Terapia di supporto
Trattamento per il tipo-specifico
Ci sono trattamenti specifici per la maggior parte delle forme di amiloidosi, anche se alcune terapie sono in fase di sperimentazione. Per tutte le forme di amiloidosi sistemica, misure di supporto possono aiutare ad alleviare i sintomi e a migliorare la qualità della vita.
Terapia di supporto
Le misure di supporto sono rivolte al sistema di organo colpito:
Renale: i pazienti con sindrome nefrotica e edema devono essere trattati con sale e restrizione dei liquidi, e diuretici; a causa della perdita di proteine in corso, l'assunzione di proteine non deve essere limitata. Il trapianto di rene è un'opzione quando il processo patologico sotteso di base è controllato, e in grado di fornire la sopravvivenza a lungo termine paragonabile a quello di altre malattie renali.
Cardiaco: i pazienti con cardiomiopatia devono essere trattati con sale, restrizione dei liquidi e diuretici dell'ansa. Altri farmaci per l'insufficienza cardiaca, tra cui digossina, ACE-inibitori, calcio-antagonisti, e beta-bloccanti sono mal tollerati e controindicati. Il trapianto di cuore ha avuto successo in pazienti accuratamente selezionati con amiloidosi primaria o amiloidosi da transtiretina (ATTR) e grave coinvolgimento cardiaco. Per prevenire recidive nel cuore trapiantato, i pazienti con amiloidosi primaria devono essere sottoposti a chemioterapia aggressiva diretta al disturbo clonale delle plasmacellule e i pazienti con polineuropatia o cardiomiopatia amiloide da transtiretina (ATTR) sintomatica legata alla transtiretina devono essere valutati per avviare terapie antitranstiretina.
Gastrointestinale: i pazienti con diarrea possono trarre beneficio dalla loperamide. Quelli con sazietà precoce e ritenzione gastrica possono beneficiare di metoclopramide.
Sistema nervoso: nei pazienti con neuropatia periferica, gabapentin, pregabalin o duloxetina possono alleviare il dolore.
L'ipotensione ortostatica spesso migliora con alte dosi di midodrina; questo farmaco può causare ritenzione urinaria nei maschi più anziani, ma la complicanza farmacologica dell'ipertensione supina è raramente un problema in questa popolazione. Anche le calze elastiche possono aiutare, e il fludrocortisone in pazienti senza edema periferico, anasarca, o insufficienza cardiaca. Nei pazienti con ipotensione ortostatica refrattaria, possono essere aggiunti la midodrina, il fludrocortisone o la droxidopa.
Amiloidosi primaria
Per l'amiloidosi primaria:
Il tempestivo inizio della terapia cellulare è essenziale per preservare la funzione degli organi e prolungare la vita.
La maggior parte dei farmaci utilizzati per il mieloma multiplo è stata utilizzata per l'amiloidosi primaria; la scelta del farmaco, la dose, e il calendario spesso devono essere modificati quando la funzione di alcuni organi è compromessa.
La chemioterapia con un agente alchilante (p. es., melfalan, ciclofosfamide) in combinazione con corticosteroidi è stato il primo regime a dimostrare qualche beneficio. Alte dosi di melfalan EV, in combinazione con il trapianto autologo di cellule staminali, possono essere molto efficaci in pazienti selezionati (1).
Anche l'inibitore del proteasoma bortezomib e immunomodulanti (p. es., lenalidomide) possono risultare efficaci. Uno studio sull'anticorpo monoclonale daratumumab più ciclofosfamide, bortezomib e desametasone in pazienti con amiloidosi AL nuovamente diagnosticata (esclusi quelli con insufficienza cardiaca di classe NYHA III e IV, proteina natriuretica N-terminale pro-B-type [NTproBNP] > 8500 pg/mL [> 1005 pmol/L] ed eGFR < 20 mL/minuto/m2) hanno mostrato un inedito alto tasso di risposta ematologica (2). La risposta ematologica si basa sui livelli di proteine monoclonali sierici e urinari determinati dall'elettroforesi con immunofissazione e dai livelli di catene leggere sieriche con rapporti kappa/lambda. Tuttavia, i dati di sopravvivenza a lungo termine mancano.
Tutti i trattamenti disponibili hanno come obiettivo le cellule B clonali o le plasmacellule nell'amiloidosi AL. Sono in corso studi sugli anticorpi antifibril, come il birtamimab e il CAEL-101 (3).
L'amiloidosi primaria localizzata può essere trattata con radioterapia a basso dosaggio in quanto le plasmacellule sono altamente radiosensibili.
Amiloidosi da accumulo di transtiretina (ATTR)
Per l'amiloidosi da accumulo di transtiretina (ATTR):
Trapianto di fegato
Farmaci stabilizzatori del tetramero
Farmaci per il silenziamento genico
Il trapianto di fegato, che sostituisce la sede primaria di produzione della proteina mutante con un nuovo organo che produce normale transitretina, può essere efficace soprattutto in alcune mutazioni della TTR se eseguito all'esordio della malattia (neuropatia precoce e senza coinvolgimento cardiaco). Il trapianto più tardi nel corso della malattia spesso porta a una cardiomiopatia e a una neuropatia amiloide progressiva a causa dell'errato ripiegamento e del deposito della proteina transtiretrina wild-type su depositi di amiloide preesistenti.
Numerosi farmaci hanno dimostrato di stabilizzare i tetrameri di transtiretina che circola nel plasma, inibendo il malripiegamento della transtiretina e la formazione di fibrille e rallentando effettivamente la progressione della malattia neurologica, preservando la qualità della vita. Questi stabilizzatori della transtiretina comprendono il diflunisal, un farmaco antinfiammatorio generico ampiamente disponibile, e il tafamidis (4, 5).
Il silenziamento del gene TTR, utilizzando RNA anti-senso o "RNA interference" per bloccare la traduzione del TTR in mRNA, riduce efficacemente i livelli sierici di transtiretina e migliora gli esiti neurologici in circa il 50% dei pazienti, e sembra capace di riparare i nervi lesi in alcuni pazienti (6, 7). Sono disponibili i farmaci di silenziamento genico, il patisiran, l'inoteren e il vutrisiran.
Un trial del vutrisiran, un silenziatore genico di seconda generazione, ha dimostrato un miglioramento dei risultati funzionali in pazienti con polineuropatia amiloide familiare (8). I dati preliminari di un altro studio suggeriscono che i silenziatori genici possono essere efficaci nel trattamento della cardiomiopatia nei pazienti con amiloidosi da transtiretina (ATTR) (9).
Amiloidosi da transtiretina wild type (ATTRwt)
Per l'amiloidosi da transtiretina wild type (ATTRwt):
Farmaci stabilizzatori del tetramero
La stabilizzazione della transtiretina utilizzando il tafamidis, in pazienti con cardiomiopatia amiloide legata alla transtiretina (ATTR o ATTRwt), ha dimostrato di ridurre la mortalità per tutte le cause e le ospedalizzazioni per cause cardiovascolari (5). Sono in corso studi clinici che esaminano l'effetto dei silenziatori del gene TTR sulla cardiomiopatia nei pazienti con amiloidosi da transtiretina wild type (ATTRwt), così come nella cardiomiopatia in pazienti con amiloidosi da transtiretina (ATTR) caratterizzata dalla proteina mutante (10).
A differenza dell'amiloidosi da transitiretina (ATTR) ereditaria, il trapianto di fegato non è efficace per i pazienti con amiloidosi causata da transtiretina wild type perché la proteina amiloidogenica è un transtiretina strutturalmente normale.
Amiloidosi secondaria
Nell'amiloidosi AA causata dalla febbre familiare mediterranea, la colchicina orale aè efficace.
Per altri tipi di amiloidosi secondaria, il trattamento è diretto all'infezione sottostante, alla malattia infiammatoria, o al cancro.
I farmaci colchicina, anti-IL1, anti-IL6 o anti-TNF possono essere usati per interrompere i segnali indotti dalle citochine, diminuendo il processo infiammatorio che guida la produzione epatica di amiloide sierica A.
Riferimenti relativi al trattamento
1. Sanchorawala V, Sun F, Quillen K, et al: Long-term outcome of patients with AL amyloidosis treated with high-dose melphalan and stem cell transplantation: 20-year experience. Blood 126: 2345–2347, 2015. doi: 10.1182/blood-2015-08-662726
2. Kastritis E, Palladini G, Minnema MC, et al: Daratumumab-Based Treatment for Immunoglobulin Light-Chain Amyloidosis. N Engl J Med 385(1):46-58, 2021. doi:10.1056/NEJMoa2028631
3. Quarta CC, Fontana M, Damy T, et al: Changing paradigm in the treatment of amyloidosis: From disease-modifying drugs to anti-fibril therapy. Front Cardiovasc Med 9:1073503, 2022. doi:10.3389/fcvm.2022.1073503
4. Berk JL, Suhr OB, Obici L, et al: Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA 310: 2658–2667, 2013. doi: 10.1001/jama.2013.283815
5. Maurer MS, Schwartz JH, Gundapaneni B, et al: Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med 379:1007–1016, 2018.
6. Adams D, Gonzalez-Duarte A, O'Riordan WD, et al: Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med 379:11–21, 2018.
7. Benson MD, Waddington-Cruz M, Berk JL, et al: Inotersen treatment for patients with transthyretin amyloidosis. N Engl J Med 379:22–31, 2018.
8. Adams D, Tournev IL, Taylor MS, et al: Efficacy and safety of vutrisiran for patients with hereditary transthyretin-mediated amyloidosis with polyneuropathy: a randomized clinical trial. Amyloid 30(1):1-9, 2023. doi:10.1080/13506129.2022.2091985
9. Maurer MS, Fontanta MA, Berk JL, et al: Primary results from APOLLO-B, a phase 3 study of patisiran in patients with transthyretin-mediated amyloidosis with cardiomyopathy. Abstract presented at International Symposium of Amyloidosis, September 2022,Heidelberg Germany.
10. Writing Committee, Kittleson MM, Ruberg FL, et al: 2023 ACC Expert Consensus Decision Pathway on Comprehensive Multidisciplinary Care for the Patient With Cardiac Amyloidosis: A Report of the American College of Cardiology Solution Set Oversight Committee [published correction appears in J Am Coll Cardiol 81(11):1135, 2023]. J Am Coll Cardiol 81(11):1076-1126, 2023. doi:10.1016/j.jacc.2022.11.022
Prognosi dell'amiloidosi
La prognosi dipende dal tipo di amiloidosi e dagli apparati interessati ma, con la terapia e le cure specifiche ed appropriate, molti pazienti hanno un'aspettativa di vita eccellente.
L'amiloidosi primaria complicata da grave cardiomiopatia ha la prognosi più severa, con una sopravvivenza mediana < 1 anno. Nei casi di amiloidosi da transtiretina (ATTR) non trattati di solito si arriva alla patologia cardiaca e neurologica terminale nell'arco di 5-15 anni. L'amiloidosi da transtiretina wild type (ATTRwt) ha tipicamente la più lenta progressione rispetto alle altre forme di amiloidosi sistemica che coinvolgono il cuore; tuttavia, i pazienti con amiloidosi da transtiretina wild type (ATTRwt) passano a insufficienza cardiaca sintomatica e decedono entro una mediana di 4 anni dalla diagnosi bioptica.
La prognosi dell'amiloidosi secondaria dipende in gran parte dall'efficacia del trattamento del disturbo sottostante infettivo, infiammatorio o neoplastico.
Punti chiave
L'amiloidosi è un gruppo di malattie in cui alcune proteine mal ripiegate si aggregano in fibrille insolubili che si depositano all'interno di organi, causando disfunzioni.
Molte proteine differenti sono inclini al malripiegamento; alcune di queste proteine sono prodotte da un difetto genetico o da determinate patologie, mentre altre coinvolgono catene leggere di immunoglobuline prodotte da plasmacellule monoclonali o da altre malattie linfoproliferative a cellule B.
La proteina amiloidogenica determina il tipo di amiloide e il decorso clinico della malattia, anche se le manifestazioni cliniche dei diversi tipi possono sovrapporsi.
Molti organi possono essere colpiti, ma il coinvolgimento cardiaco comporta una prognosi particolarmente nefasta; la cardiomiopatia da amiloidosi comporta normalmente disfunzione diastolica, insufficienza cardiaca e blocco cardiaco o aritmia.
La diagnosi è con biopsia; il tipo di amiloidosi è determinato da una serie di test immunologici, genetici e biochimici. La spettrometria di massa è il metodo più sensibile e specifico per la tipizzazione dell'amiloide.
Un'adeguata terapia di supporto aiuterà ad alleviare i sintomi e a migliorare la qualità della vita; il trapianto di organi può aiutare in pazienti selezionati.
Trattare il processo sottostante; per l'amiloidosi primaria causata da plasmacellule o per le malattie linfoproliferative, la chemioterapia può essere molto efficace; per l'amiloidosi secondaria, i farmaci antinfiammatori e gli antibiotici possono risultare utili.
Per l'amiloidosi da transtiretina (ATTR) ereditaria, le terapie stabilizzanti a piccole molecole e i farmaci di silenziamento genico inibiscono o potenzialmente invertono il deterioramento neurologico; per i pazienti con cardiomiopatia amiloide (ATTR o ATTRwt), il tafamidis riduce la mortalità per tutte le cause e i ricoveri per malattie cardiovascolari.







