


Le miositi autoimmuni sono caratterizzate da alterazioni infiammatorie e degenerative dei muscoli (polimiosite, miopatia immuno-mediata necrotizzante) o di cute e muscoli (dermatomiosite). Le manifestazioni comprendono ipostenia simmetrica, occasionalmente dolorabilità e sostituzione fibrosa del muscolo, a volte con atrofia, principalmente dei muscoli prossimali dei cingoli. La diagnosi si basa sui reperti clinici e sulle anomalie nei test muscolari, che possono comprendere test della creatinchinasi, RM, elettromiografia e biopsia muscolare. Diversi tipi di miosite hanno manifestazioni polmonari e cardiache. La terapia è a base di corticosteroidi associati ad immunosoppressori e/o immunoglobuline EV.
La miosite autoimmune è più comune nelle donne rispetto agli uomini in un rapporto 2:1. L'incidenza è 3-4 volte maggiore nelle persone di colore rispetto ai bianchi. Queste malattie possono insorgere a ogni età, ma si manifestano più comunemente tra i 40 e i 60 anni o, nei bambini, tra i 5 e i 15 anni.
Eziologia della miosite autoimmune
L'eziologia della miosite autoimmune sembra essere legata a una reazione autoimmune all'interno del tessuto muscolare nei soggetti geneticamente suscettibili. Si verifica un raggruppamento familiare, e i sottotipi dell'antigene leucocitario umano sono associati alla miosite. Per esempio, gli alleli dell'aplotipo ancestrale 8.1 (HLA-DRB1*03-DQA1*05-DQB1*02) aumentano il rischio di polimiosite, dermatomiosite e di malattia polmonare interstiziale. Una miosite di natura virale e una neoplasia maligna sottostante possono rappresentare possibili eventi scatenanti. L'associazione di un tumore maligno con la dermatomiosite (meno con la polimiosite) suggerisce che una neoplasia maligna possa favorire la miosite in seguito a una reazione autoimmune contro un antigene comune, presente nel muscolo e nel tumore.
Fisiopatologia della miosite autoimmune
Le alterazioni anatomopatologiche comprendono danno e atrofia cellulare, con gradi variabili di infiammazione. I muscoli di mani, piedi e viso sono colpiti in misura minore rispetto ad altri muscoli scheletrici. Il coinvolgimento dei muscoli della faringe, dell'esofago superiore e occasionalmente del cuore può compromettere la funzione di questi organi. Soprattutto in pazienti con anticorpi antisintetasi può innescarsi un processo infiammatorio a livello articolare e polmonare.
La dermatomiosite è caratterizzata da deposizione di immunocomplessi nei vasi ed è considerata una vasculite complemento-mediata. Al contrario, la polimiosite è caratterizzata da un danno muscolare diretto mediato dalle cellule T, mentre le miopatie immuno-mediate necrotizzanti sono caratterizzate da infiltrati dove predominano macrofagi e miopagocitosi.
Classificazione della miosite autoimmune
La miosite autoimmune può essere classificata in 4 gruppi, principalmente basati su istopatologia e manifestazione clinica:
Polimiosite
Dermatomiosite
Miopatie immuno-mediate necrotizzanti
Miosite da corpi inclusi
La dermatomiosite può essere distinta dalla polimiosite per la presenza di caratteristici reperti cutanei di dermatomiosite (vedi Sintomatologia). Anche l'istopatologia muscolare differisce. La dermatomiosite e la polimiosite possono manifestarsi come malattie muscolari pure o come parte della sindrome da antisintetasi che possono essere associati ad artrite (di solito non progressiva), febbre, malattia polmonare interstiziale, ipercheratosi della porzione radiale delle dita (mani da meccanico), e sindrome di Raynaud.
Le miopatie immuno-mediate necrotizzanti il più delle volte comprendono la miosite correlata agli anticorpi del segnale di riconoscimento delle particelle e la miosite indotta da statine, di solito hanno una presentazione aggressiva, livelli di creatinchinasi molto elevati, e non coinvolgono organi extramuscolari (1).
La miosite da inclusione del corpo provoca debolezza dei muscoli prossimali delle gambe, ma spesso coinvolge i muscoli distali (p. es., i muscoli della mano e del piede) spesso con atrofia muscolare. Si sviluppa in età più avanzata, ha una progressione più lenta e generalmente non risponde alla terapia immunosoppressiva.
La miosite autoimmune può inoltre sovrapporsi ad altre malattie reumatiche autoimmuni, p. es., lupus eritematoso sistemico, sclerosi sistemica, connettivite mista. Questi pazienti presentano sintomatologia dei disturbi di sovrapposizione oltre alla miosite (manifesti come dermatomiosite o polimiosite).
Riferimenti relativi alla classificazione
1. Lundberg IE, Fujimoto M, Vencovsky J, et al. Idiopathic inflammatory myopathies. Nat Rev Dis Primers 7(1):86, 2021. doi:10.1038/s41572-021-00321-x
Sintomatologia della miosite autoimmune
L'insorgenza della miosite autoimmune può essere acuta (in particolare nei bambini) o subdola (particolarmente negli adulti). Possono anche comparire poliartralgie, sindrome di Raynaud, disfagia, sintomi polmonari (p. es., tosse, dispnea), e disturbi sistemici (soprattutto febbre, astenia e perdita di peso). La malattia grave è caratterizzata da disfagia, disfonia e/o debolezza diaframmatica.
L'ipostenia muscolare può progredire nel corso di settimane o mesi. Tuttavia, è necessario che il 50% delle fibre muscolari venga distrutto per causare debolezza sintomatica (ossia, la debolezza muscolare indica una miosite in stadio avanzato). I pazienti possono avere difficoltà a sollevare le braccia al di sopra delle spalle, a salire le scale e ad assumere la stazione eretta da seduti. A volte si sviluppano dolorabilità e atrofia del muscolo. I pazienti possono richiedere l'uso di una sedia a rotelle o rimanere a letto a causa della debolezza dei muscoli pelvici e dei cingoli scapolari. Anche i muscoli flessori del collo possono essere colpiti gravemente, causando incapacità di sollevare la testa dal cuscino. Il coinvolgimento dei muscoli faringei e dell'esofago superiore può compromettere la deglutizione e predisporre all'aspirazione. I muscoli delle mani, dei piedi e del viso non sono coinvolti fatta eccezione per la miosite da corpi inclusi, in cui è caratteristico il coinvolgimento distale, specialmente delle mani. Raramente, si sviluppano retrazioni degli arti.
Le manifestazioni articolari comprendono poliartralgie o poliartropatie, associate a tumefazione e ad altre caratteristiche delle artropatie non deformanti. Insorgono in genere in un sottogruppo di pazienti con positività per anticorpi anti-Jo-1 o altri anticorpi antisintetasi.
Il coinvolgimento viscerale (tranne che della faringe e dell'esofago superiore) è meno frequente nella miosite autoimmune che in altre malattie reumatiche (p. es., lupus eritematoso sistemico, sclerosi sistemica). Occasionalmente, e specialmente nei pazienti con anticorpi antisintetasi, una malattia polmonare interstiziale (caratterizzata da dispnea e tosse) rappresenta la manifestazione clinica più rilevante. Possono verificarsi coinvolgimento cardiaco, soprattutto disturbi della conduzione e disfunzione ventricolare. I sintomi gastrointestinali, più frequenti nei bambini, sono dovuti a una vasculite associata e possono comprendere ematemesi, melena e perforazione intestinale ischemica.
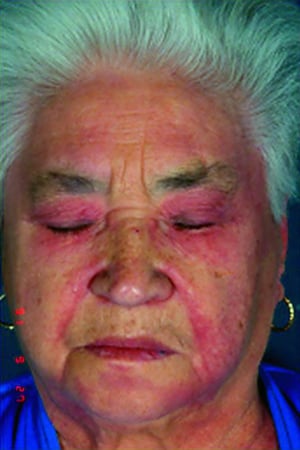
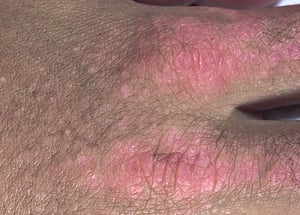
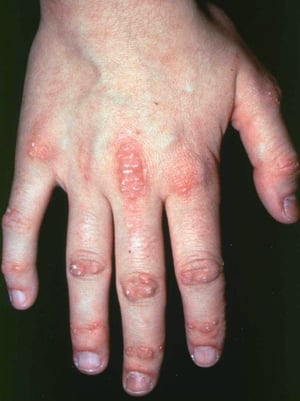
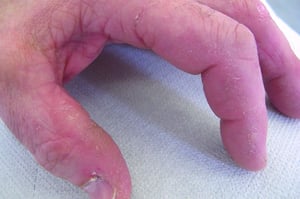
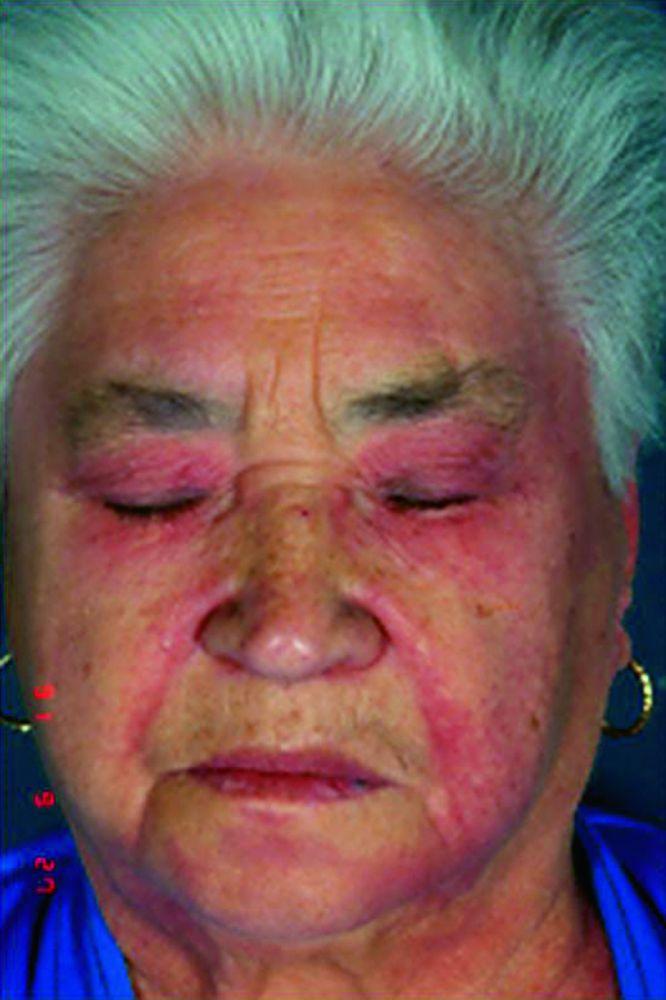
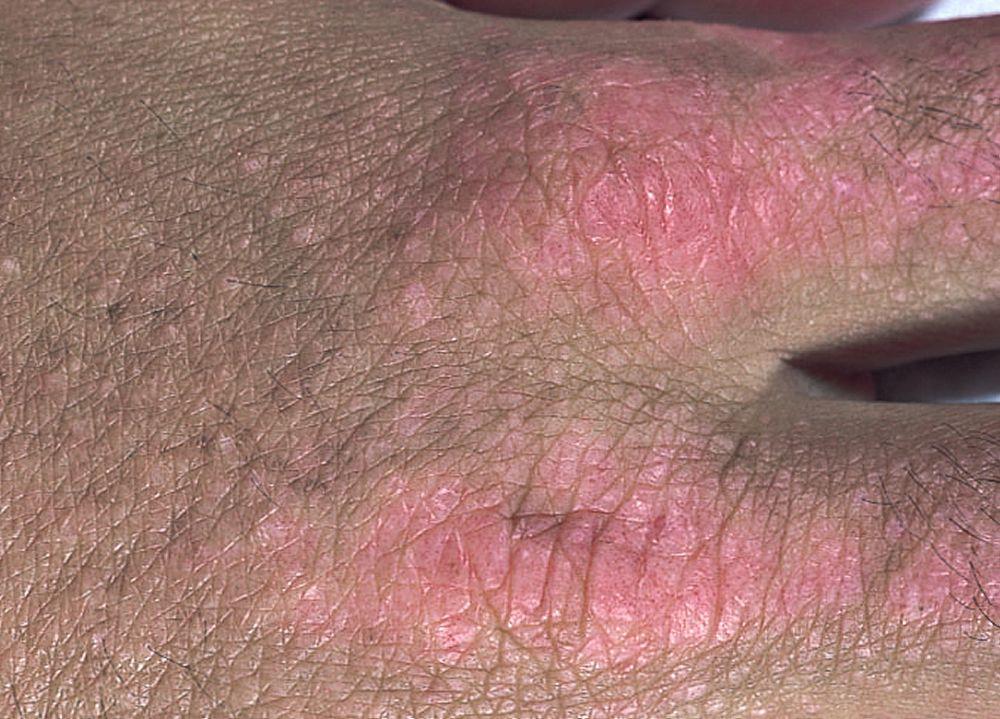
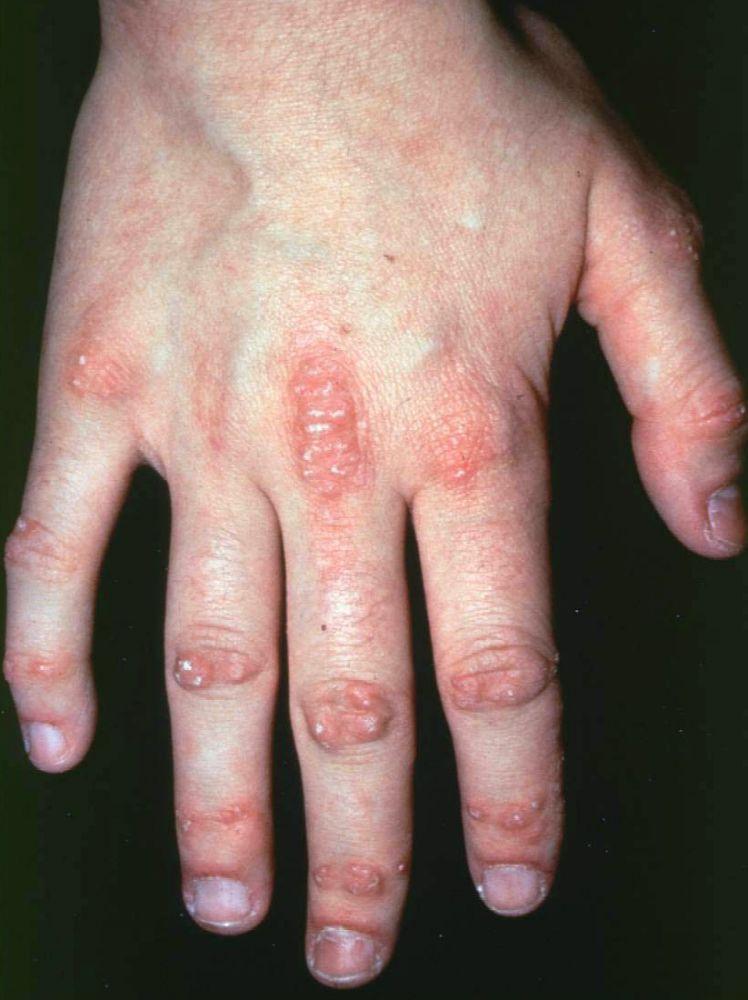
Le manifestazioni cutanee, che compaiono nella dermatomiosite, tendono a essere scure ed eritematose. La fotosensibilità e l'ulcerazione cutanea sono visibili. L'edema periorbitale dall'aspetto violaceo (rash eliotropo) è relativamente specifico per dermatomiosite. In altre sedi corporee, il rash può essere leggermente rilevato e liscio o squamoso; esso può apparire sulla fronte, sulla "V" del collo e sulle spalle, sul torace, sulla schiena, sugli avambracci, sulle gambe, sulla parte laterale delle cosce, sui gomiti e sulle ginocchia, sui malleoli mediali e sul dorso delle articolazioni interfalangee prossimali e metacarpofalangee (papule di Gottron, una lesione relativamente specifica). La base e i lati delle unghie possono essere iperemici o ispessiti. La dermatite desquamante con fissurazioni della cute può estendersi sul versante radiale delle dita. Può verificarsi calcificazione sottocutanea e muscolare, in particolare nei bambini. Le lesioni cutanee primarie spesso scompaiono completamente, ma possono essere seguite da alterazioni secondarie (p. es., pigmentazione brunastra, atrofia, neovascolarizzazione persistente, cicatrizzazione). Il rash sul cuoio capelluto può sembrare psoriaforme ed essere intensamente pruriginoso.
I cambiamenti caratteristici della pelle possono verificarsi in assenza di malattia muscolare, nel qual caso la malattia è chiamata dermatomiosite amiopatica.
© Springer Science+Business Media
© Springer Science+Business Media
© Springer Science+Business Media
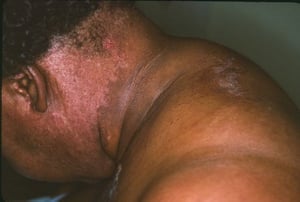
Image courtesy of Karen McKoy, MD.
© Springer Science+Business Media

© Springer Science+Business Media

© Springer Science+Business Media

© Springer Science+Business Media

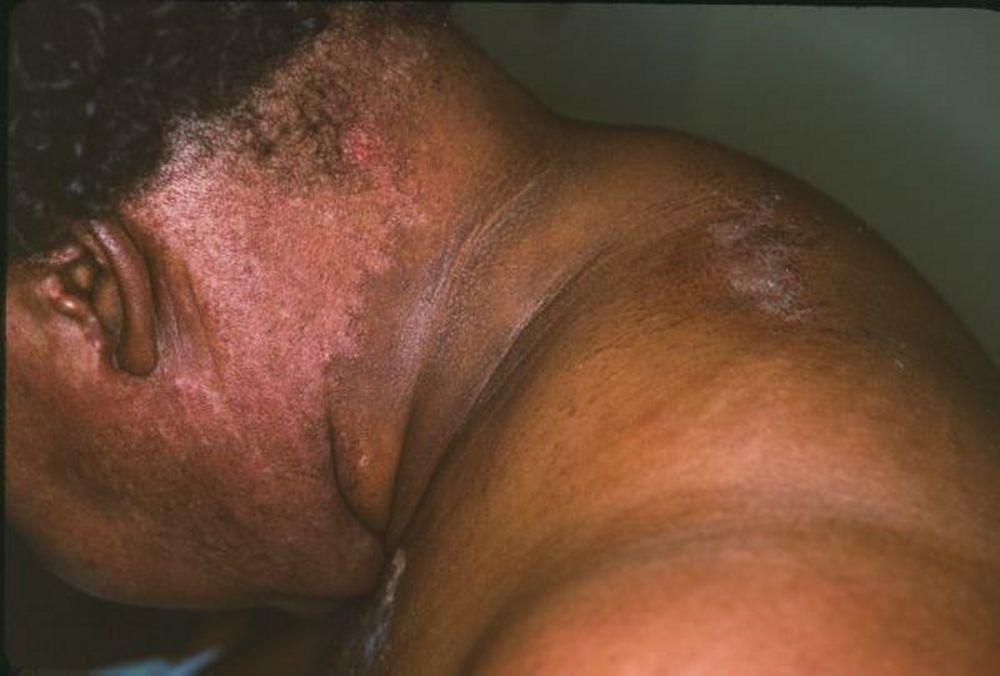
Image courtesy of Karen McKoy, MD.

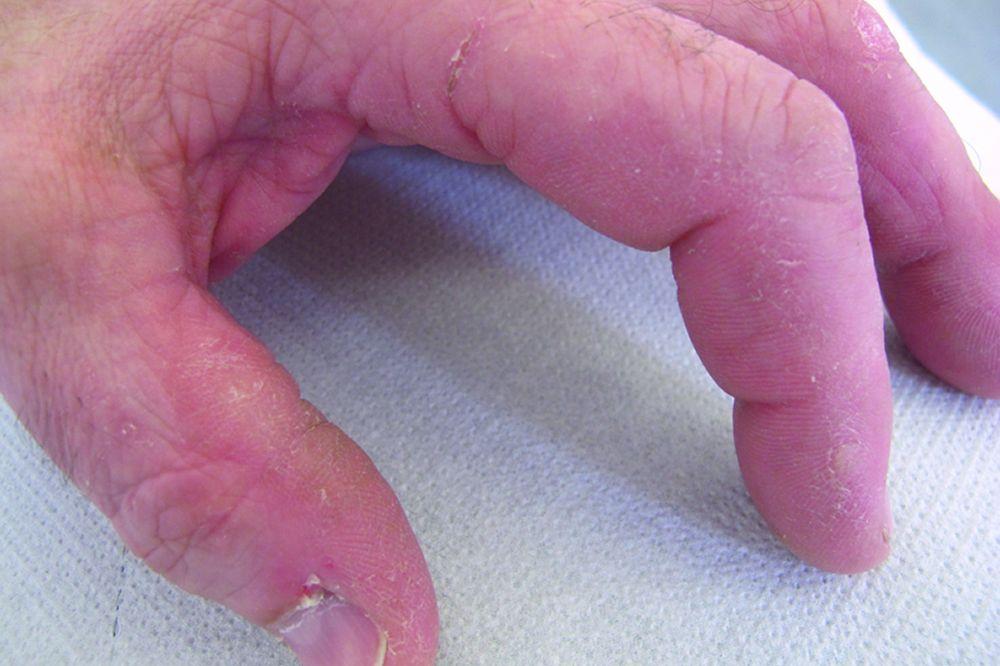
© Springer Science+Business Media
Diagnosi della miosite autoimmune
Criteri clinici
Biopsia muscolare (dirimente)
La miosite autoimmune deve essere sospettata in pazienti con ipostenia dei muscoli prossimali con o senza dolore muscolare. La dermatomiosite invece si deve sospettare in pazienti con sintomi di miosite e reperti cutanei compatibili con dermatomiosite. La diagnosi di miosite autoimmune richiede la positività del maggior numero dei seguenti 5 criteri:
Ipostenia dei muscoli prossimali
Rash caratteristico
Innalzamento degli enzimi muscolari (se la creatinchinasi [CK] non è elevata, aminotransferasi o aldolasi [che sono meno specifiche della CK])
Alterazioni muscolari caratteristiche all'esame elettromiografico o alla RM
Alterazioni all'esame bioptico muscolare (esame diagnostico dirimente)
I reperti della biopsia possono variare, ma l'infiammazione cronica con la degenerazione e una certa rigenerazione muscolare sono peculiari. La polimiosite e la dermatomiosite possono spesso essere distinte tramite biopsia muscolare. Si consiglia, prima di intraprendere il trattamento della polimiosite, di effettuare diagnosi di certezza attraverso la biopsia muscolare per escludere altre patologie muscolari, come quelle dovute a enzimi mancanti o difettosi, miosite necrotizzante e rabdomiolisi post-virale. La biopsia muscolare non è solitamente necessaria quando i reperti cutanei sono caratteristici della dermatomiosite. Non vi è alcun riscontro patognomico della cute per la dermatomiosite alla biopsia, ma l'assenza di immunofluorescenza diretta aiuta a distinguere l'eruzione dall'eruzione in pazienti con lupus eritematoso sistemico.
Per aumentare la sensibilità del risultato della biopsia, il campione bioptico deve essere ottenuto da un muscolo che ha una o più delle seguenti caratteristiche:
Debolezza all'esame clinico
Edema muscolare individuato attraverso la RM
Coppia controlaterale di un muscolo del quale è stata documentata un'alterazione all'esame elettromiografico
Gli esami di laboratorio possono orientare nel sospetto diagnostico per questa malattia, determinarne la gravità, identificarne la sovrapposizione con altre patologie e contribuire a rilevare complicanze. Si devono eseguire i test specifici per la ricerca degli auto-Ac. Gli anticorpi antinucleo (ANA) sono positivi fino all'80% dei pazienti con dermatomiosite e polimiosite. Se il test per gli anticorpi antinucleo (ANA) è positivo, ulteriori test per tipi specifici di anticorpi sono importanti per aumentare il sospetto di una sindrome sovrapposta.
Il decorso e le manifestazioni cliniche sono associate a particolari anticorpi come descritto nella tabella Autoanticorpi nella miosite autoimmune. La relazione tra questi auto-anticorpi e la patogenesi della malattia resta incerta, benché l'anticorpo anti-Jo-1 sia un marker significativo di alveolite fibrosante, fibrosi polmonare, artrite e sindrome di Raynaud. Non ci sono anticorpi specifici per la polimiosite.
L'evidenza di un aumento del rischio di cancro è relativamente forte nella dermatomiosite e meno forte per la polimiosite. Pertanto, lo screening oncologico deve essere considerato per i pazienti ≥ 40 anni affetti da dermatomiosite o per i pazienti ≥ 60 anni affetti da polimiosite in quanto tali pazienti hanno spesso tumori misconosciuti. Lo screening deve comprendere almeno esame obiettivo che includa seni, pelvi e retto (con il test per la ricerca del sangue occulto nelle feci); emocromo con formula; profilo biochimico; mammografia; analisi delle urine; RX torace; e ogni altro esame appropriato per l'età del paziente.
Indagini supplementari devono basarsi sull'anamnesi e sui risultati dell'esame obiettivo. Alcune autorità mediche raccomandano di eseguire una TC del torace, addome e pelvi, così come una colonscopia, in particolare nei pazienti affetti da dermatomiosite. Non è necessario sottoporre a screening i pazienti più giovani che non presentano sintomi di neoplasie maligne.
Prognosi della miosite autoimmune
Remissioni a lungo termine (addirittura evidenti guarigioni) si verificano in una percentuale di pazienti che arriva fino al 50% entro 5 anni, in genere nei bambini. In qualsiasi momento tuttavia possono comparire ricadute. Il tasso complessivo di sopravvivenza a 5 anni è del 75%, ed è più elevato nei bambini.
Il decesso negli adulti è preceduto da grave e progressiva ipostenia muscolare, disfagia, denutrizione, polmonite ab ingestis o insufficienza respiratoria con sovrapposizione infettiva.
Il decesso nei bambini con dermatomiosite può essere la conseguenza di una vasculite intestinale.
La dermatomiosite e la polimiosite sono state associate a un aumento del rischio di cancro. Una neoplasia maligna, quando presente, influenza generalmente la prognosi complessiva.
Trattamento della miosite autoimmune
Corticosteroidi
Immunosoppressori (p. es., metotrexato, azatioprina, micofenolato mofetile, rituximab, tacrolimus)
Immunoglobuline EV
L'attività fisica deve essere moderatamente ridotta fino a quando l'infiammazione non regredisce.
I corticosteroidi sono inizialmente i farmaci di elezione. Per la malattia in fase acuta agli adulti si somministra prednisone per via orale 1 mg/kg (solitamente all'incirca da 40 a 60 mg) 1 volta/die. Per le malattie gravi con disfagia o debolezza del muscolo respiratorio, il trattamento di solito inizia con terapia con corticosteroidi a dosi elevate (p. es., metilprednisolone da 0,5 a 1 g EV 1 volta/die per 3-5 giorni).
Ripetute determinazioni della creatinchinasi sono il migliore indicatore di efficacia terapeutica. Tuttavia, in pazienti affetti da atrofia muscolare diffusa, i livelli a volte sono normali nonostante sia presente una miosite cronica in fase attiva. I risultati RM dell'edema muscolare o elevati livelli di creatininchinasi generalmente differenziano una miosite in fase di riacutizzazione da una miopatia indotta da corticosteroidi. L'aldolasi è un'alternativa, essendo meno specifica per il danno muscolare rispetto alla creatinfosfochinasi, ma a volte può essere positiva nei pazienti con miosite e livelli normali di creatinfosfochinasi. Quando i livelli degli enzimi scendono o raggiungono valori normali in molti pazienti in 6-12 settimane, a seguito di un miglioramento della forza muscolare, la dose di corticosteroidi può essere gradualmente ridotta. Se i livelli degli enzimi muscolari aumentano di nuovo, la dose di corticosteroidi è solitamente aumentata in attesa dell'effetto completo di altri farmaci.
L'obiettivo generale è quello di eliminare rapidamente l'infiammazione e di minimizzare l'esposizione a lungo termine ai corticosteroidi, motivo per cui un secondo farmaco (tipicamente metotrexato, tacrolimus o azatioprina come farmaci non-steroidei di prima linea) viene iniziato contemporaneamente ai corticosteroidi o poco dopo in modo che il prednisone possa essere ridotto a una dose massima di 5 mg/die, idealmente entro circa 6 mesi. Le immunoglobuline EV sono una buona opzione per i pazienti che non rispondono rapidamente alla terapia, sviluppano complicanze infettive con corticosteroidi ad alto dosaggio e altri immunosoppressori o sottoposti a chemioterapia. Alcuni esperti possono utilizzare una combinazione di tutte e 3 le terapie nei casi gravi o quando è presente tossicità da corticosteroidi. Ai bambini è necessario somministrare dosi iniziali di prednisone da 30 a 60 mg/m2 1 volta/die.
Talvolta i pazienti trattati cronicamente con dosi elevate di corticosteroidi divengono progressivamente deboli dopo la risposta iniziale per il sovrapporsi di una miopatia da corticosteroidi indolore. In questi pazienti, la creatinfosfochinasi rimane normale anche se i pazienti sono astenici.
La miosite associata a cancro è più refrattaria ai corticosteroidi. La miosite associata a neoplasia può andare incontro a remissione se il tumore viene asportato.
Le persone con una malattia autoimmune sono a più alto rischio di aterosclerosi e devono essere attentamente monitorate. I pazienti in terapia a lungo termine con corticosteroidi devono ricevere la terapia di profilassi per l'osteoporosi. Se viene utilizzata una terapia immunosoppressiva combinata, i pazienti devono ricevere una profilassi per le infezioni opportunistiche, come quelle da Pneumocystis jirovecii (vedi prevenzione della polmonite da Pneumocystis jirovecii), e i vaccini contro le infezioni comuni (p. es., polmonite streptococcica, influenza, COVID-19).
Punti chiave
La debolezza muscolare causata dalla miosite è molto spesso prossimale.
Le eruzioni cutanee eliotropiche e le papule Gottron sono specifiche per la dermatomiosite.
Per stabilire la diagnosi, bisogna cercare il rash cutaneo caratteristico, la debolezza muscolare, elevati livelli di creatinchinasi, e le alterazioni muscolari all'esame elettromiografico o RM.
Se i pazienti non presentano i caratteristici reperti cutanei, eseguire la biopsia muscolare per confermare la diagnosi.
Si consideri lo screening nei pazienti ≥ 40 anni con dermatomiosite e nei pazienti di età ≥ 60 anni con polimiosite per il rischio di cancro.
Trattare i pazienti con corticosteroidi e con altri immunosoppressori.







