




I difetti congeniti degli arti comprendono la presenza, alla nascita, di arti mancanti, incompleti, in sovrannumero, o anormalmente sviluppati.
(Vedi anche Introduzione ai disturbi cranio-facciali e muscoloscheletrici congeniti.)
Difetti in riduzione degli arti
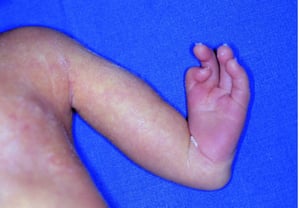
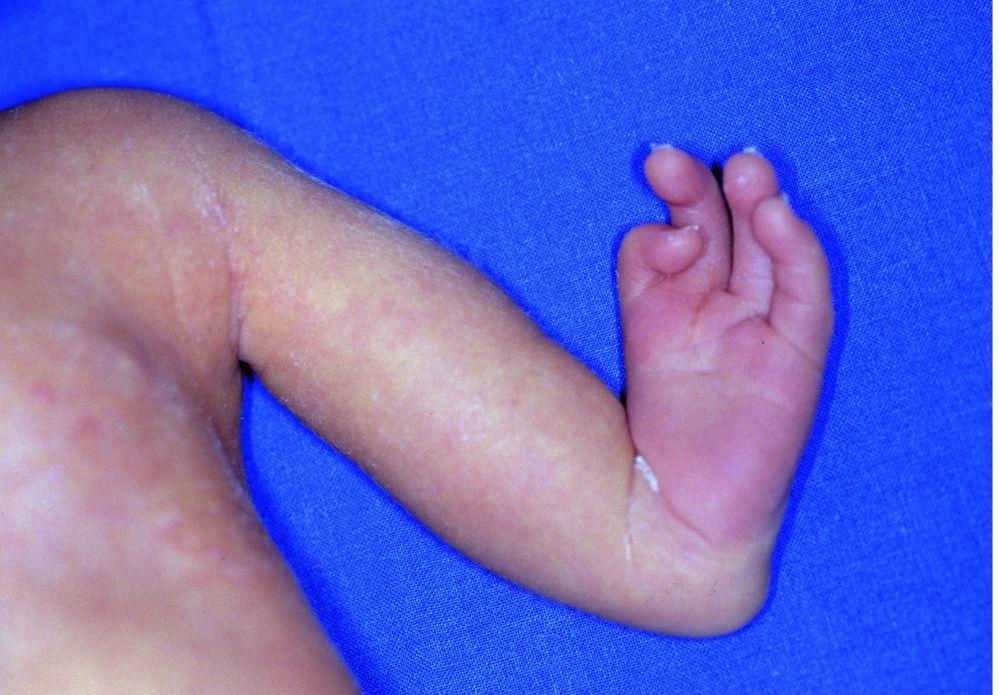
Nelle amputazioni e nei difetti in riduzione congeniti gli arti sono assenti o incompleti alla nascita. La prevalenza totale è di 7,9/10 000 nati vivi. Nella maggior parte dei casi sono dovuti ad inibizione primitiva dello sviluppo intrauterino o a un disturbo conseguente a distruzione intrauterina di tessuto embrionale normale. Le estremità superiori sono più comunemente colpite.
I difetti congeniti in riduzione degli arti hanno molte cause e si verificano spesso come componenti di varie sindromi congenite. Agenti teratogeni (p. es., talidomide, vitamina A) sono cause note di ipoplasia/assenza di arti. La causa più comune di amputazione congenita degli arti è rappresentata da difetti dei tessuti molli e/o disturbi vascolari, come i difetti di arti da briglia amniotica, in cui tralci fibrosi di amnion imbrigliano o si fondono con tessuti fetali.
I difetti in riduzione degli arti possono essere
Longitudinali (più frequenti)
Trasversali
I difetti longitudinali comprendono specifiche alterazioni di sviluppo (p. es., assenza completa o parziale del radio, del perone o della tibia). L'ipoplasia del radio è l'anomalia più frequente degli arti superiori, e l'ipoplasia del perone è l'anomalia più frequente degli arti inferiori. Circa due terzi dei casi sono associati ad altre malattie congenite, tra cui la sindrome di Adams-Oliver (aplasia congenita della cute con aplasia parziale delle ossa del cranio e difetto terminale trasversale degli arti), la sindrome di Holt-Oram, la sindrome TAR (trombocitopenia-absent radius), l'anemia di Franconi e la sindrome VACTERL (vertebral anomalies, anal atresia, cardiac malformations, tracheoesophageal fistula, renal anomalies and radial aplasia, and limb [arti] anomalies).
© Springer Science+Business Media
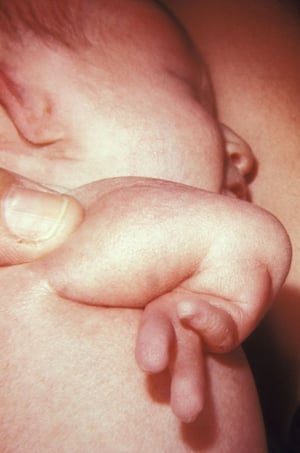
Image courtesy of CDC/Dr. James W. Hanson via the Public Health Image Library of the Centers for Disease Control and Prevention.
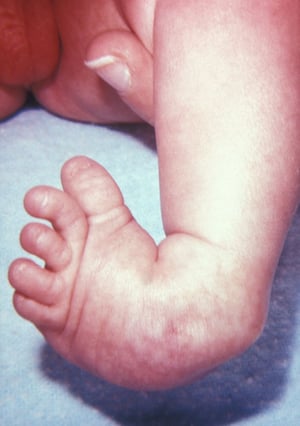
Image courtesy of CDC/Dr. James W. Hanson via the Public Health Image Library of the Centers for Disease Control and Prevention.

© Springer Science+Business Media

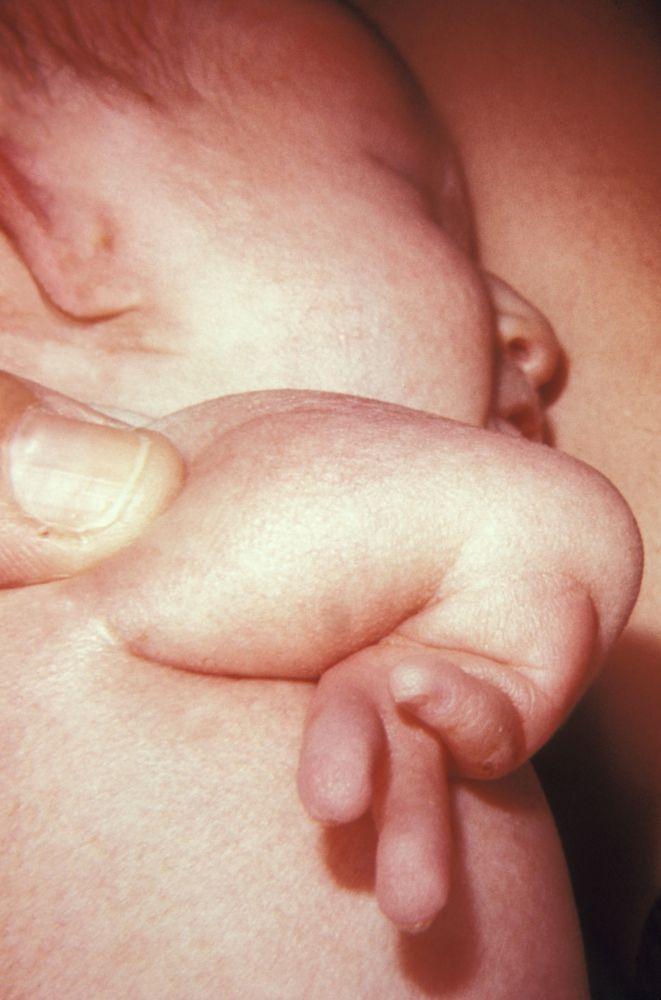
Image courtesy of CDC/Dr. James W. Hanson via the Public Health Image Library of the Centers for Disease Control and Prevention.

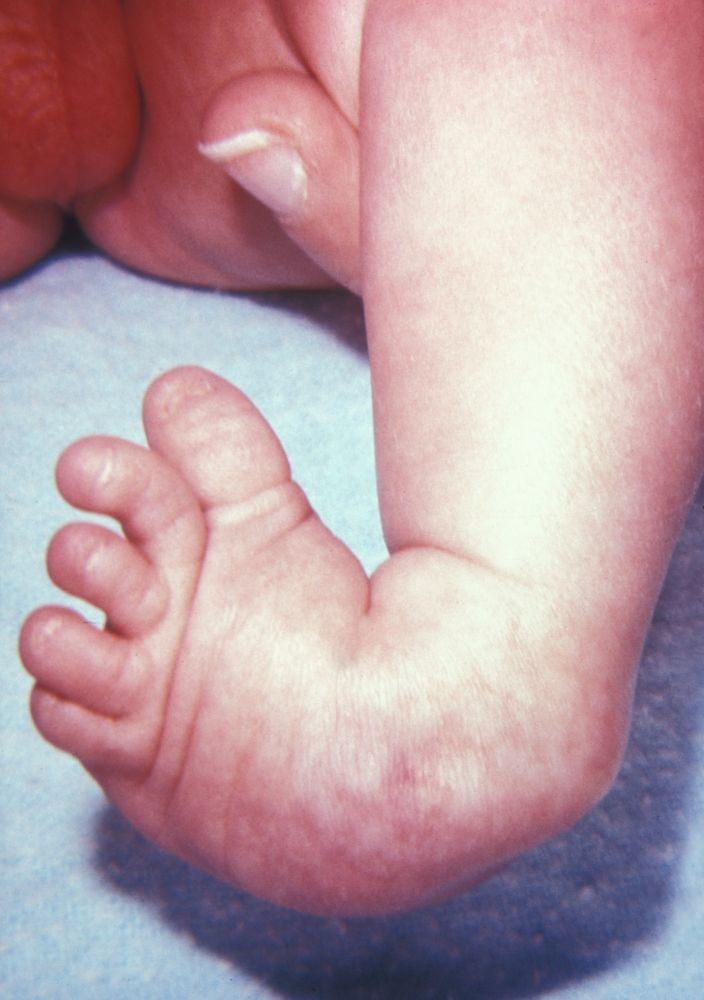
Image courtesy of CDC/Dr. James W. Hanson via the Public Health Image Library of the Centers for Disease Control and Prevention.
Nei difetti trasversali, sono assenti tutti gli elementi oltre un certo livello e gli arti sono ridotti a monconi. Le bande amniotiche sono la causa più comune; la gravità del difetto varia in base alla localizzazione della banda amniotica e, in genere, non ci sono altri difetti o anomalie. I casi rimanenti fanno per lo più parte di sindromi genetiche come la sindrome di Adams-Oliver o anomalie cromosomiche.
In presenza di difetti trasversali o longitudinali, a seconda dell'eziologia, i bambini possono anche avere ossa ipoplasiche o bifide, sinostosi, duplicazioni, lussazioni o altri difetti ossei; per esempio, nel deficit focale prossimale del femore, la porzione prossimale del femore e l'acetabolo non si sviluppano. Possono essere interessati uno o più arti e il tipo di difetto può essere diverso in ciascun arto. Le malformazioni del sistema nervoso centrale sono rare.
Polidattilia
La polidattilia consiste nella presenza di dita sovrannumerarie ed è la più frequente anomalia congenita degli arti. Questa anomalia è classificata come preassiale, centrale e postassiale.
Nella polidattilia preassiale vi è un pollice o un alluce in sovrannumero. Le manifestazioni vanno da un allargamento o duplicazione della falange distale a una duplicazione completa del dito. L'anomalia si può presentare in forma isolata, eventualmente con trasmissione autosomica dominante, o può essere parte di alcune sindromi genetiche, inclusa la sindrome acrocallosa (con ritardo di sviluppo e difetti del corpo calloso), le sindromi di Carpenter e Pfeiffer (con craniosinostosi), le anemie di Fanconi e Diamond-Blackfan, e la sindrome di Holt-Oram (con difetti cardiaci congeniti).

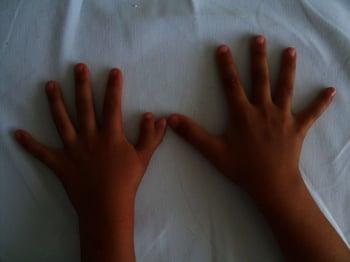
La polidattilia centrale è rara e consiste nella duplicazione del dito anulare, medio, indice. Può essere associata a sindattilia e a schisi della mano. La maggior parte dei casi è sindromica.
La polidattilia postassiale è la più frequente e consiste in un dito sovrannumerario sul lato ulnare/peroneale dell'arto. Più comunemente, il dito extra è rudimentale, ma può essere completamente sviluppato. Nelle persone di discendenza africana, questo tipo di polidattilia è di solito un difetto isolato. In altre popolazioni, viene in genere associata a una sindrome polimalformativa o ad alterazioni cromosomiche. Tra le sindromi da considerare vi sono la sindrome di cefalopolisindattilia di Greig, la sindrome di Meckel, la sindrome di Ellis-van Creveld, la sindrome di McKusick-Kaufman, la sindrome di Down, e la sindrome di Bardet-Biedl.
Sindattilia
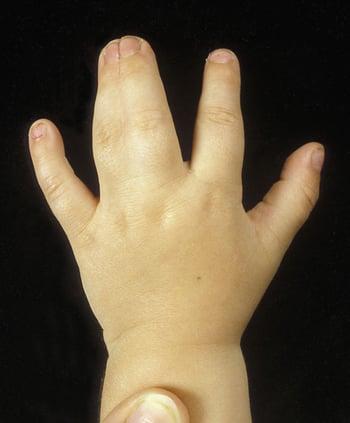
La sindattilia consiste in una membrana tra le dita delle mani o dei piedi, o nella loro fusione. Ne sono stati definiti diversi tipi, e la maggior parte segue un modello di ereditarietà autosomica dominante. Nella sindattilia semplice la fusione interessa solo i tessuti molli, mentre nella sindattilia complessa vi è anche fusione delle ossa. Sindattilia complessa è presente nella sindrome di Apert (con craniosinostosi). Sindattilia del 4o e 5o dito delle mani è comune nella displasia oculo-dento-digitale. La sindrome di Smith-Lemli-Opitz si manifesta con sindattilia del 2o e 3o dito dei piedi ed altre anomalie congenite multiple.

Diagnosi delle alterazioni congenite dell'arto
Di solito raggi X
In alcuni casi test genetico
Tipicamente, le RX sono eseguite per scoprire quali ossa sono interessate. Quando i difetti sembrano essere familiari o se si sospetta una sindrome genetica, la valutazione deve includere anche un esame approfondito per ricercare altre anomalie fisiche, cromosomiche e genetiche. Se disponibile, è utile la valutazione da parte di un genetista clinico.
Un'analisi cromosomica con microarray, dei test genici specifici o un panel di geni più ampio deve essere considerato nella valutazione dei pazienti con anomalie cranio-facciali congenite. Se i risultati di questi test non sono diagnostici, è raccomandata l'analisi dell'intero sequenziamento dell'esoma.
Trattamento delle alterazioni congenite dell'arto
Procedure chirurgiche
Dispositivi protesici
Il trattamento consiste in procedure chirurgiche per polidattilia e sindattilia. Il trattamento per gli arti mancanti o ipoplastici avviene principalmente attraverso l'uso di dispositivi protesici, che sono più utili per le mancanze degli arti inferiori e per gli arti superiori completamente o quasi completamente assenti. A prescindere dalla gravità della malformazione, se il braccio o la mano conservano un qualunque grado di funzionalità, va accertata accuratamente la loro capacità funzionale prima di consigliare l'uso di una protesi o l'intervento chirurgico. L'amputazione terapeutica dell'arto colpito o di una parte di esso deve essere presa in considerazione soltanto dopo aver valutato le implicazioni funzionali e psicologiche della perdita dell'arto e quando l'amputazione risulti necessaria per il posizionamento della protesi.
La protesi dell'arto superiore deve essere tale da poter assolvere al maggior numero possibile di funzioni, in modo che il numero dei dispositivi sia minimo. Il bambino riesce a usare nel modo più efficace la protesi quando la si monti precocemente, così che possa diventare parte integrante del suo corpo durante gli anni dello sviluppo. Gli apparecchi usati durante l'infanzia devono essere il più possibile semplici e duraturi (p. es., un uncino piuttosto che un braccio bioelettrico).
Con un efficace trattamento ortopedico e di supporto, la maggior parte dei bambini con amputazioni congenite conduce una vita normale.







