




La polmonite interstiziale linfocitaria è un'infiltrazione linfocitaria dell'interstizio alveolare e degli spazi aerei. La causa è sconosciuta. La sintomatologia comprende la tosse, la dispnea progressiva e i crepitii. La diagnosi si basa su anamnesi, esame obiettivo, diagnostica per immagini e biopsia polmonare. Il trattamento è a base di corticosteroidi, farmaci citotossici, o entrambi, sebbene l'efficacia non sia nota. La prognosi è in gran parte sconosciuta e limitata a serie di casi.
La polmonite interstiziale linfocitaria è una rara polmonite interstiziale idiopatica caratterizzata dall'infiltrazione degli alveoli e dei setti alveolari da parte di piccoli linfociti e di un numero variabile di plasmacellule. Possono essere presenti granulomi non necrotizzanti, non ben definiti, ma rari e di non facile osservazione.
Nei bambini HIV positivi la polmonite interstiziale linfocitaria è la causa più frequente di patologia polmonare dopo l'infezione da Pneumocystis ed è la malattia che si associa caratteristicamente all'AIDS fino a metà dei bambini HIV positivi. La polmonite interstiziale linfocitaria colpisce < 1% degli adulti con o senza infezione da HIV. Le donne e le ragazze sono colpite più frequentemente.
Si suppone che la causa sia una malattia autoimmune o una risposta aspecifica all'infezione da virus di Epstein-Barr, HIV o da parte di un altro virus. La prova di un'eziologia autoimmunitaria comprende la frequente associazione con la sindrome di Sjögren e altre patologie (p. es., lupus eritematoso sistemico, artrite reumatoide, tiroidite di Hashimoto).
La prova di un'eziologia virale indiretta comprende la frequente associazione con stati di immunodeficienza (HIV/AIDS, immunodeficienza combinata variabile, agammaglobulinemia) e la presenza del DNA del virus di Epstein-Barr e dell'RNA dell'HIV nel tessuto polmonare di pazienti affetti da polmonite interstiziale linfocitaria. Secondo questa ipotesi, la polmonite interstiziale linfocitaria è un'estrema manifestazione della normale capacità del tessuto linfoide polmonare di rispondere ad antigeni inalati o circolanti.
Sintomatologia della polmonite interstiziale linfocitaria
Negli adulti, la polmonite interstiziale linfocitaria provoca dispnea e tosse progressive. Queste manifestazioni progrediscono nell'arco di mesi o, in alcuni casi, di anni e compaiono all'età media di 54 anni. Si verificano perdita di peso, febbre, artralgia e sudorazione notturna, ma meno frequentemente.
L'esame obiettivo può rilevare crepitii. Reperti come epatosplenomegalia, artrite e linfoadenopatia sono infrequenti e indicano una patologia concomitante o una diagnosi alternativa.
Diagnosi della polmonite interstiziale linfocitaria
TC ad alta risoluzione
Per la conferma, biopsia
La diagnosi di polmonite interstiziale linfocitaria si sospetta generalmente in pazienti a rischio con sintomi compatibili. Vengono effettuati imaging e talvolta biopsia polmonare.
La RX torace mostra opacità lineari bibasali reticolari o nodulari, un reperto aspecifico che si ritrova in molte infezioni polmonari. Nelle fasi più avanzate della malattia possono essere presenti opacità alveolari, cisti, o entrambe.
Viene eseguita una TC ad alta risoluzione del torace che permette di stabilire l'estensione della patologia, definire l'anatomia ilare e identificare il coinvolgimento pleurico. I quadri TC ad alta risoluzione sono altamente variabili. I reperti caratteristici sono i noduli centrolobulari e subpleurici, un ispessimento della trama vascolare, le opacità a vetro smerigliato nodulari, e le aree cistiche.

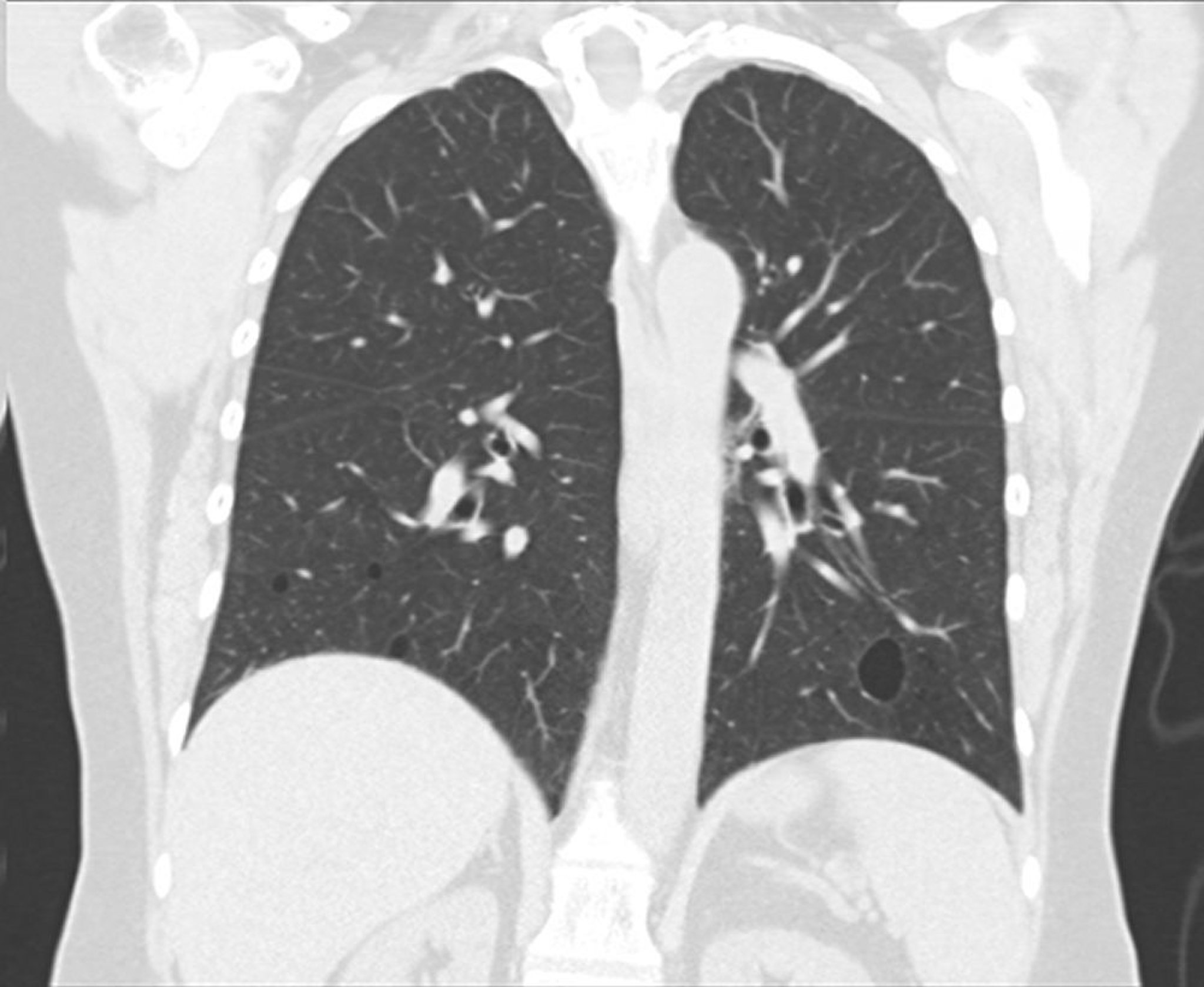
This image courtesy of Tami Bang, MD.
Può essere presente una marcata ipossiemia.
Il lavaggio broncoalveolare può essere eseguito per escludere un'infezione e può rilevare un aumento del numero dei linfociti.
I test di laboratorio di routine e l'elettroforesi delle proteine sieriche vengono effttuati perché circa l'80% dei pazienti presenta un'anomalia delle proteine sieriche, più comunemente una gammopatia policlonale e un'ipogammaglobulinemia, il significato delle quali non è noto. Altri test di laboratorio (p. es., sierologie, livelli di immunoglobuline, test per l'HIV) vengono effettuati per identificare le potenziali cause di polmonite interstiziale linfocitaria secondaria.
La biopsia polmonare con dimostrazione dell'ispessimento dei setti alveolari con infiltrati formati dovuti a linfociti e ad altre cellule immunitarie (plasmacellule, immunoblasti, istiociti) è richiesta per la diagnosi negli adulti. Gli infiltrati compaiono occasionalmente lungo i bronchi e i vasi, ma più comunemente lungo i setti alveolari. Sul tessuto occorre eseguire le colorazioni immunoistochimiche e la citometria a flusso per distinguere la polmonite interstiziale linfocitaria dai linfomi primitivi. Nella polmonite interstiziale linfocitaria, l'infiltrato è policlonale (formato sia da cellule T sia da B), mentre altri linfomi producono infiltrati monoclonali. Altri segni frequenti sono centri germinali e cellule giganti multinucleate con granulomi non caseosi.
Trattamento della polmonite interstiziale linfocitaria
Corticosteroidi o farmaci citotossici
Il trattamento della polmonite interstiziale linfocitaria è con corticosteroidi, farmaci citotossici o entrambi, anche se come per molte altre cause di malattia polmonare interstiziale, l'efficacia di tale approccio non è nota.
Prognosi della pneumopatia interstiziale linfocitaria
L'evoluzione naturale e la prognosi della polmonite interstiziale linfocitaria negli adulti sono poco note. Si può verificare una risoluzione spontanea, una risoluzione dopo il trattamento con corticosteroidi o altri farmaci immunosoppressivi, una progressione verso un linfoma o lo sviluppo di una fibrosi polmonare con insufficienza respiratoria. La sopravvivenza a cinque anni è del 50-66%. Frequenti cause di morte sono l'infezione, lo sviluppo di un linfoma maligno (5%) e la fibrosi progressiva.
Punti chiave
La polmonite interstiziale linfocitica è rara nel complesso, ma è uno dei disturbi polmonari più comuni nei bambini HIV-positivi.
La sintomatologia tende a essere aspecifica.
Eseguire TC ad alta risoluzione, lavaggio broncoalveolare e talvolta biopsia polmonare.
Trattare i pazienti con corticosteroidi, farmaci citotossici o entrambi.







