


La connectivite mixte est un syndrome rare qui se caractérise par les manifestations cliniques du lupus érythémateux disséminé, de la sclérodermie, de la polymyosite/dermatomyosite et de la polyarthrite rhumatoïde et par des taux très élevés d'anticorps antinucléaires circulants dirigés contre un antigène de type ribonucléoprotéine (RNP). Une tuméfaction de la main, un syndrome de Raynaud, des polyarthralgies, une myopathie inflammatoire, une hypomotilité œsophagienne et une maladie pulmonaire interstitielle sont fréquents. Le diagnostic repose sur l'association des signes cliniques, par la présence d'anticorps antiribonucléoprotéines et par l'absence d'anticorps spécifiques d'autres maladies auto-immunes. Le traitement varie selon la gravité des maladies et l'atteinte d'organes mais comprend habituellement des corticostéroïdes et également des immunosuppresseurs.
La connectivite mixte est cosmopolite et touche toutes les races avec un pic d'incidence à l'adolescence et à la vingtaine. Environ 80% de personnes qui ont cette maladie sont des femmes. La cause de la connectivite mixte est inconnue. Chez de nombreux patients, la maladie évolue vers une sclérodermie classique ou vers un lupus érythémateux disséminé.
Symptomatologie de la connectivite mixte
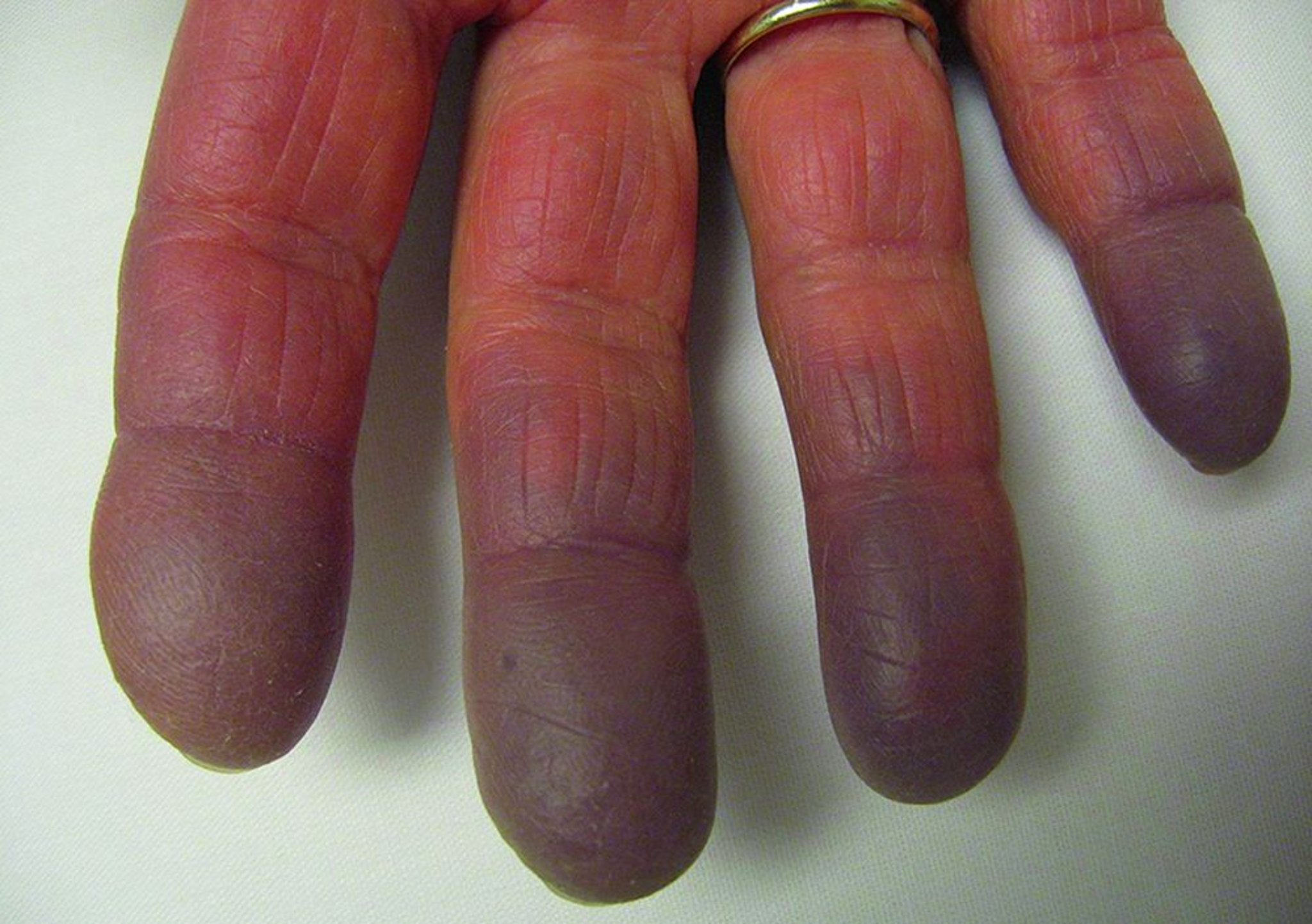
Un syndrome de Raynaud (phénomène de Raynaud) peut précéder les autres manifestations. Fréquemment, les premières manifestations ressemblent à celles d'un lupus érythémateux disséminé, d'une sclérodermie, d'une polymyosite ou même d'une polyarthrite rhumatoïde. De nombreux patients, semblent avoir initialement une maladie du tissu conjonctif mixte ou indifférenciée. Les manifestations de la maladie peuvent progresser et s'étendre, et leur expression clinique se modifie avec le temps.
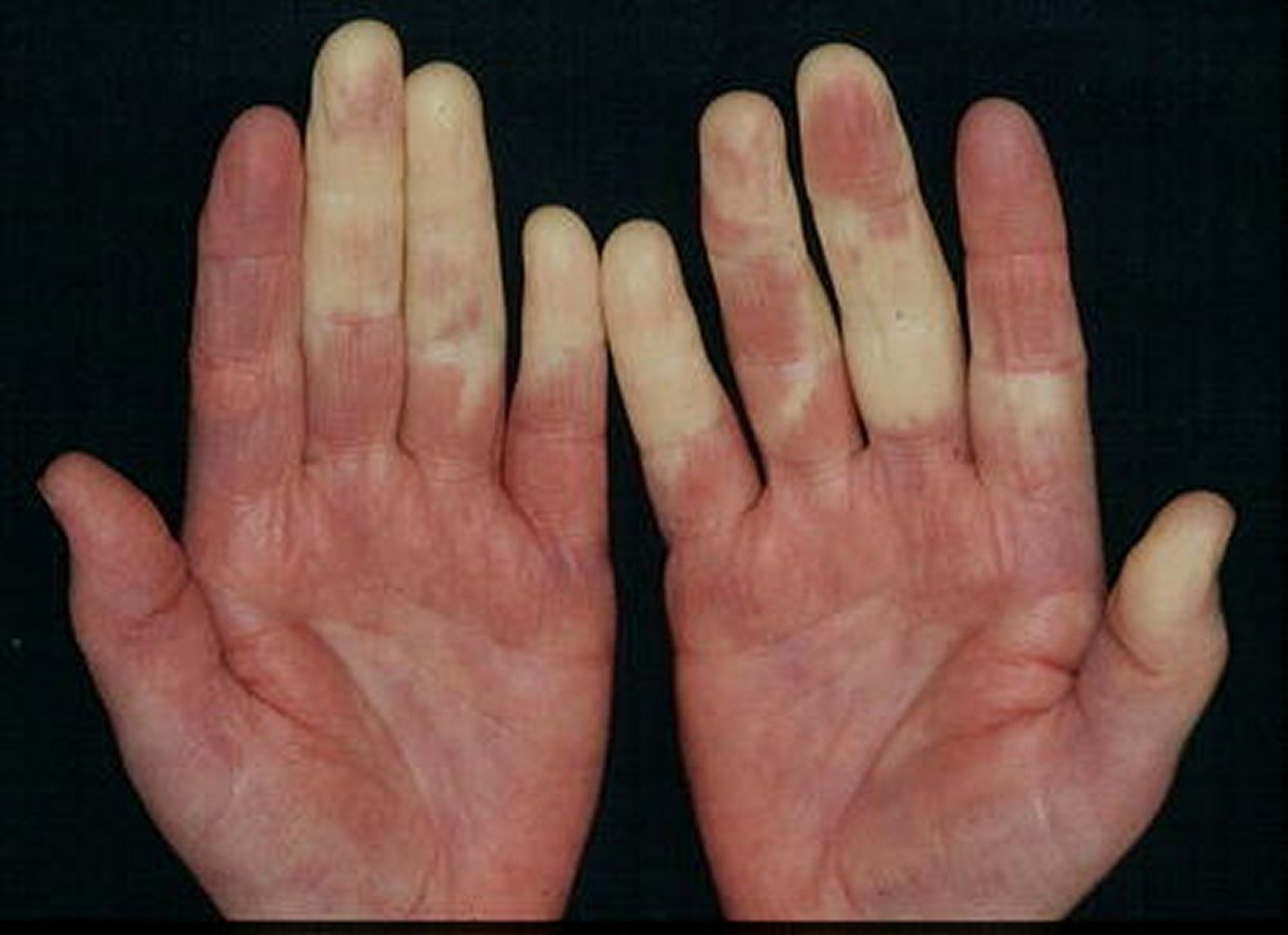
La tuméfaction initiale diffuse des mains est typique, mais non universelle. Les signes cutanés comprennent des éruptions ressemblant à celles du lupus ou d'une dermatomyosite. Des lésions diffuses de type sclérodermique et les nécroses ou ulcérations ischémiques de la pulpe des doigts peuvent se développer.

© Springer Science+Business Media

© Springer Science+Business Media
Presque tous les patients ont des polyarthralgies et 75% ont des arthrites. Souvent, l'arthrite n'est pas déformante, mais des érosions et des déformations similaires à la polyarthrite rhumatoïde sont parfois observées (p. ex., déformations en boutonnière et en col-de-cygne). Une faiblesse musculaire proximale, avec ou sans douleur à la palpation, est habituelle, typiquement chez les personnes qui ont des niveaux élevés d'enzymes musculaires (p. ex., creatine kinase [CK]).
L'atteinte rénale (le plus souvent néphropathie membraneuse) se produit chez environ 25% des patients et est généralement légère; une atteinte sévère, avec une morbidité ou une mortalité, est atypique dans la connectivite mixte. Les poumons sont affectés dans jusqu'à 75% des cas de connectivite mixte. La pneumopathie interstitielle du poumon est la manifestation la plus fréquente; l'hypertension artérielle pulmonaire est une cause majeure de décès. Une insuffisance cardiaque peut survenir. Un syndrome de Sjögren peut se développer.
Diagnostic de la connectivite mixte
Dosage des anticorps antinucléaires (AAN), des anticorps contre les antigènes extractibles (anticorps anti-ribonucléoprotéine U1 ou RNP) et des anticorps anti-Smith (Sm) et les anticorps anti-ADN
Atteinte d'un organe déterminée selon les indications cliniques
Une connectivite mixte doit être suspectée lorsque les patients ont des signes compatibles avec un lupus érythémateux disséminé, une sclérodermie ou une polymyosite.
Des tests pour les AAN et les antigènes U1 RNP sont pratiqués en premier. Presque tous les patients présentent des titres élevés d'anticorps antinucléaires (AAN) mouchetés. Les anticorps anti-U1 RNP sont habituellement présents à des taux très élevés. Les anticorps contre la composante Sm ribonucléase-résistante de l'antigène nucléaire extractible (anticorps anti-Sm) et contre l'ADN double-brin (négatifs dans la connectivite mixte par définition) sont mesurés pour exclure d'autres troubles. La présence d'anticorps anti-RNP n'est pas suffisante pour poser le diagnostic de connectivite mixte; des signes cliniques typiques sont également nécessaires.
Les facteurs rhumatoïdes sont souvent positifs et les titres peuvent être élevés. La VS est souvent élevée.
L'hypertension artérielle pulmonaire doit être détectée le plus tôt possible par des épreuves fonctionnelles respiratoires et une échocardiographie. Un bilan plus approfondi dépend de symptomatologie; des manifestations de la myosite, une atteinte rénale ou des signes pulmonaire justifient d'autres examens complémentaires.
La créatine kinase, l'IRM, l'électromyogramme ou la biopsie musculaire peuvent permettre de confirmer la présence d'une myosite dans le cadre de la connectivite mixte.
Pronostic de la connectivite mixte
La survie globale à 10 ans est d'environ 80% mais le pronostic dépend largement des manifestations prédominantes. Les patients qui présentent des caractéristiques de sclérodermie et de polymyosite ont un plus mauvais pronostic. Les patients ont des risques d'athérosclérose accrus. Les causes de décès sont l'hypertension artérielle pulmonaire, l'insuffisance rénale, l'infarctus du myocarde, la perforation colique, les infections disséminées et les hémorragies cérébrales. Certains patients connaissent des rémissions pendant de nombreuses années sans traitement.
Traitement de la connectivite mixte
Anti-inflammatoires non stéroïdiens (AINS) ou antipaludéens (p. ex., hydroxychloroquine, chloroquine) en cas de maladie bénigne
Corticostéroïdes et autres immunosuppresseurs (p. ex., méthotrexate, azathioprine, mycophénolate mofétil) pour les maladies modérées à sévères
Inhibiteurs calciques (p. ex., nifédipine) et inhibiteurs de la phosphodiestérase (p. ex., tadalafil) dans le syndrome de Raynaud
La prise en charge générale et le traitement médicamenteux initial sont adaptés au problème clinique spécifique et sont similaires à ceux du lupus érythémateux disséminé ou au phénotype clinique dominant. La plupart des patients qui ont une atteinte modérée ou sévère réagissent aux corticostéroïdes, en particulier s'ils sont traités précocement, et à d'autres immunosuppresseurs (p. ex., le méthotrexate, l'azathioprine, le mycophénolate mofétil). La forme modérée est souvent contrôlée par les AINS, les antipaludéens (p. ex., hydroxychloroquine, chloroquine) ou, parfois, par des doses très faibles de corticostéroïdes. L'atteinte sévère d'un organe majeur requiert habituellement des doses plus importantes de corticostéroïdes (p. ex., prednisone 1 mg/kg par voie orale 1 fois/jour) ou d'immunodépresseurs. Si les patients développent des signes de myosite ou de sclérodermie, le traitement est le même que pour ces maladies.
Les patients qui ont un syndrome de Raynaud doivent être traités en fonction de leurs symptômes et selon la tolérance de leur pression artérielle par des inhibiteurs calciques (p. ex., la nifédipine) et des inhibiteurs de la phosphodiestérase (p. ex., le tadalafil).
L'athérosclérose doit faire l'objet d'un suivi chez les patients. Les patients sous corticostéroïdes au long cours peuvent recevoir un biphosphonate pour prévenir l'ostéoporose. Si un traitement immunosuppresseur d'association est utilisé, les patients doivent recevoir une prophylaxie contre les infections opportunistes, telles que par Pneumocystis jirovecii (voir prévention de la pneumonie à Pneumocystis jirovecii) et des vaccins contre les infections fréquentes (p. ex., pneumonie streptococcique, grippe, COVID-19).
Certains experts encouragent le dépistage périodique de l'hypertension pulmonaire par des épreuves fonctionnelles respiratoires et/ou une échocardiographie tous les 1 à 2 ans, en fonction des symptômes.
Points clés
La connectivite mixte ressemble le plus souvent au lupus érythémateux disséminé, à la sclérodermie, et/ou à la polymyosite.
Généralement, des AAN et des anticorps contre U1 RNP sont présents et les anticorps anti-Sm et anti-ADN sont absents mais la présence d'anticorps anti-RNP n'est pas suffisante pour poser le diagnostic.
Anticiper une hypertension artérielle pulmonaire.
Traiter une maladie bénigne par les AINS ou les antipaludéens et une maladie plus grave par des corticostéroïdes et d'autres immunosuppresseurs.







