




La fibrosis quística (mucoviscidosis) es una enfermedad hereditaria que causa que ciertas glándulas produzcan secreciones anormalmente espesas, lo que provoca a su vez lesiones en órganos y tejidos, especialmente en los pulmones y en el tubo digestivo.
La fibrosis quística está causada por genes anómalos heredados que alteran la función de la proteína de la fibrosis quística, lo que da lugar a secreciones espesas y pegajosas que obstruyen los pulmones y otros órganos.
Los síntomas característicos consisten en distensión abdominal, heces sueltas y poco aumento de peso, así como tos, sibilancias e infecciones respiratorias frecuentes a lo largo de la vida.
El diagnóstico se basa en los resultados de la prueba del sudor y/o estudios genéticos.
Más de la mitad de los estadounidenses que sufren esta enfermedad son adultos.
Los tratamientos consisten en antibióticos, broncodilatadores, fármacos para fluidificar las secreciones pulmonares, tratamientos de desobstrucción de las vías respiratorias para los problemas respiratorios, suplementos de enzimas pancreáticas y vitaminas para los problemas digestivos, y fármacos para mejorar la función de la proteína de la fibrosis quística en personas con ciertos genes anormales.
Algunas personas necesitan un trasplante de hígado y de pulmón.
La fibrosis quística es una enfermedad hereditaria que conduce a un acortamiento de la esperanza de vida. En Estados Unidos, por ejemplo, se presenta en 1 de cada 3300 niños de ascendencia caucásica y en 1 de cada 15 300 niños de ascendencia africana. Es poco frecuente en personas de ascendencia asiática. En Estados Unidos hay aproximadamente 40 000 personas con fibrosis quística y en todo el mundo hay 100 000 personas diagnosticadas de fibrosis quística. Dado que los avances en el tratamiento de esta enfermedad han ampliado la esperanza de vida de las personas con fibrosis quística, cerca del 60% de personas con esta enfermedad en Estados Unidos son adultos. La fibrosis quística afecta por igual a niños y niñas.
Fibrosis quística: no solo una enfermedad pulmonar
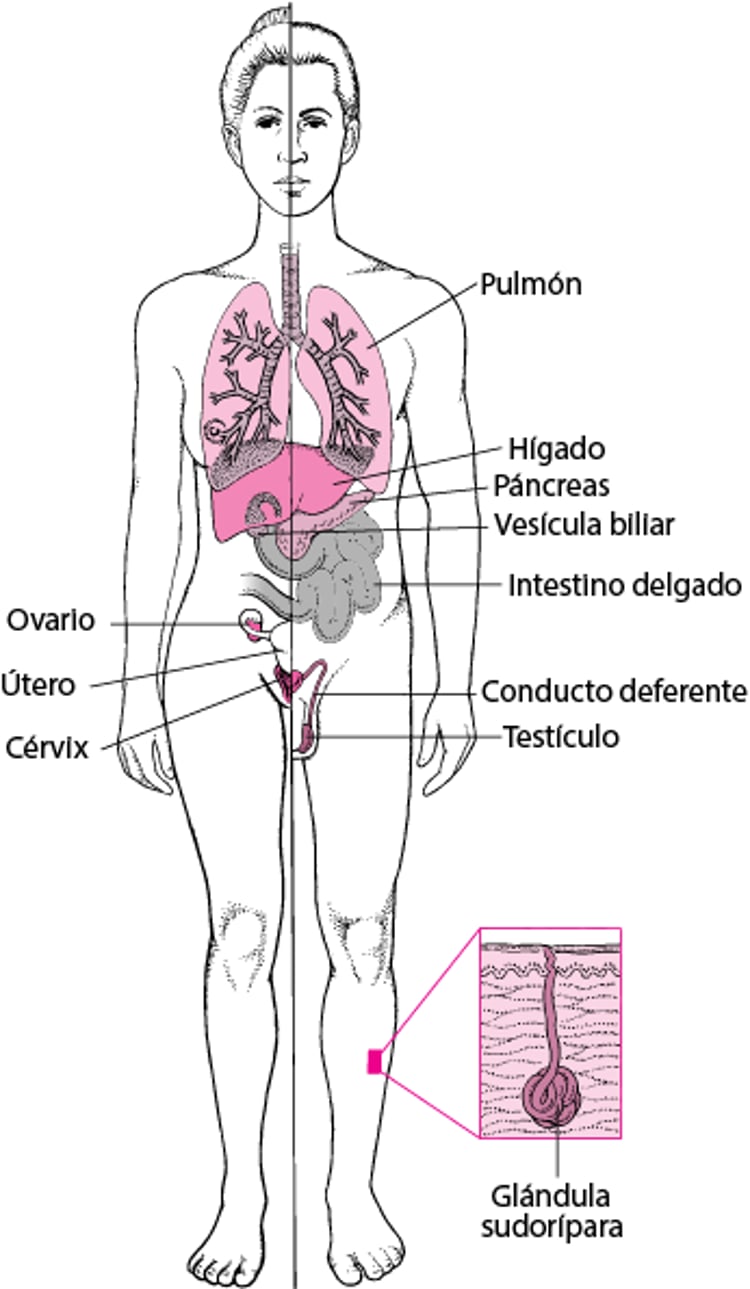
La fibrosis quística afecta a los pulmones y también a varios otros órganos. En los pulmones, las secreciones bronquiales espesas obstruyen las vías respiratorias de pequeño calibre, que se infectan e inflaman. A medida que la enfermedad progresa, las paredes bronquiales aumentan de grosor, las vías respiratorias se llenan de secreciones infectadas, algunas zonas del pulmón se contraen y los ganglios linfáticos aumentan de tamaño. En el hígado, las secreciones espesas bloquean las vías biliares. Pueden desarrollarse cálculos en la vesícula biliar. En el páncreas, las secreciones espesas pueden bloquear la glándula por completo de modo que las enzimas digestivas no pueden alcanzar el intestino. El páncreas pueden producir menos insulina, por lo que algunas personas desarrollan diabetes (por lo general en la adolescencia o en la edad adulta). En el intestino delgado, el íleo meconial (un tipo de obstrucción intestinal) puede ser resultado de secreciones espesas y puede requerir cirugía en los recién nacidos. Los genitales se ven afectados por la fibrosis quística en diversas formas, ocasionando infertilidad en personas de sexo masculino. El fluido que secretan las glándulas sudoríparas de la piel contiene más sal de lo normal. |

Causas de la fibrosis quística
Genes anómalos
La fibrosis quística se produce cuando una persona hereda dos copias defectuosas (variantes) de un determinado gen, una de cada progenitor. Este gen se llama regulador de la conductancia transmembrana de la fibrosis quística (RCFQ). Existen diversas variantes del gen CFTR. Por ejemplo, la más común se llama variante F508del. El gen CFTR (RCFQ) controla la producción de una proteína que regula el desplazamiento de cloruro, bicarbonato y sodio (sal) a través de las membranas celulares. Las variantes en el gen CFTR provocan que la proteína se vuelva disfuncional. Si la proteína no funciona correctamente, se ve interrumpido el movimiento de cloruro, bicarbonato y sodio, dando lugar al espesamiento y al aumento de la viscosidad de las secreciones de todo el organismo.
En la población mundial, alrededor de 3 de cada 100 personas de ascendencia caucásica tienen una copia defectuosa del gen CFTR. Las personas con una sola copia defectuosa del gen son portadoras de la enfermedad, pero no la sufren. Alrededor de 3 de cada 10 000 personas de ascendencia caucásica heredan dos copias defectuosas del gen y, como consecuencia, desarrollan fibrosis quística.
Secreciones anómalas
La fibrosis quística afecta muchos órganos en todo el cuerpo y casi todas las glándulas que secretan líquidos a través de un conducto (glándulas exocrinas).
Los órganos más frecuentemente afectados son
Pulmones
Páncreas
Intestinos
Hígado y vesícula biliar
Órganos reproductores
Los pulmones son normales al nacer, pero los problemas se inician en el momento en que las secreciones espesas empiezan a obstruir las vías respiratorias de pequeño calibre (taponamiento mucoso). Esta obstrucción lleva a infecciones bacterianas crónicas y la inflamación causa un daño permanente a las vías respiratorias (bronquiectasia). Estas alteraciones hacen cada vez más difícil la respiración y reducen la capacidad de los pulmones para transferir oxígeno a la sangre. Las personas afectadas pueden sufrir infecciones bacterianas frecuentes que afectan los senos paranasales.
En el páncreas, la obstrucción de los conductos del páncreas evita que las enzimas digestivas alcancen el intestino delgado. La falta de estas enzimas impide la correcta absorción de grasas, proteínas y vitaminas (malabsorción). Esta mala absorción, a su vez, deriva en carencias nutricionales y falta de crecimiento. Finalmente, el páncreas se fibrosa y ya no produce suficiente insulina, por lo que algunas personas desarrollan diabetes. Sin embargo, un pequeño porcentaje de las personas que sufren fibrosis quística y portan ciertas variantes no desarrollan problemas digestivos pancreáticos.
Los intestinos se obstruyen a causa de las secreciones espesas. Esta obstrucción es común inmediatamente después del nacimiento porque el contenido del tubo digestivo del feto (llamado meconio) es anormalmente grueso. Esta obstrucción del intestino delgado se denomina íleo meconial y cuando tiene lugar en el intestino grueso se denomina síndrome del tapón meconial. Los niños mayores y los adultos también pueden tener problemas de estreñimiento y obstrucción intestinal (síndrome de obstrucción intestinal distal).
El hígado y la vesícula biliar se pueden obstruir debido a secreciones espesas, que finalmente pueden causar cicatrices hepáticas (fibrosis). Se pueden desarrollar cálculos biliares.
Los órganos reproductores se pueden obstruir debido a secreciones gruesas, que pueden causar infertilidad. Casi todos los hombres son estériles, pero la infertilidad es mucho menos frecuente en las mujeres.
Las glándulas sudoríparas secretan líquido que contiene más sal de lo normal, aumentando el riesgo de deshidratación.
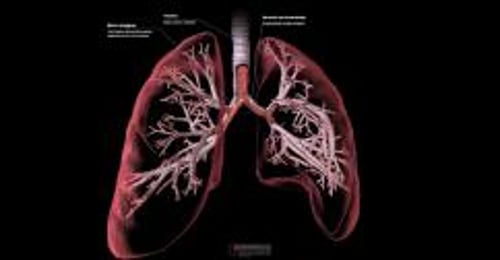
Síntomas de la fibrosis quística
Los síntomas de la fibrosis quística pueden variar dependiendo de la edad de la persona afectada.
Recién nacidos y niños pequeños
El 10% de los recién nacidos con fibrosis quística presentan íleo meconial, que causa vómitos, hinchazón (distensión) del abdomen y ausencia de deposiciones. El íleo meconial se complica a veces con una perforación del intestino, una situación peligrosa que causa infección y peritonitis (inflamación del revestimiento de la cavidad abdominal y de los órganos abdominales) y, si no se trata, choque (shock) y muerte. Algunos recién nacidos tienen una torsión del intestino sobre sí mismo (vólvulo) o un desarrollo incompleto del intestino. Los recién nacidos que presentan íleo meconial casi siempre desarrollan más adelante otros síntomas de fibrosis quística.
El primer síntoma de fibrosis quística en el niño que no presenta íleo meconial es el retraso en la recuperación del peso después de su nacimiento o el escaso aumento de peso durante las primeras 4 o 6 semanas de edad. Esta poca ganancia de peso se debe a una mala absorción de nutrientes asociada a una cantidad insuficiente de enzimas pancreáticas. El lactante tiene deposiciones frecuentes, de aspecto oleoso, abundantes y de olor desagradable y presenta un abdomen abombado (distendido). Sin tratamiento, la ganancia de peso es lenta, a pesar de un apetito normal o incluso aumentado.
Niños mayores y adultos
A menos que se haga el diagnóstico mediante una prueba de cribado neonatal, alrededor de la mitad de los niños con fibrosis quística acuden por primera vez a la consulta médica porque tienen tos, respiración sibilante e infecciones respiratorias. La tos, el síntoma más perceptible, se acompaña con frecuencia de náuseas, vómitos y alteraciones del sueño. Los niños tienen dificultad respiratoria, sibilancias o ambas. A medida que la enfermedad progresa, los niños desarrollan una disminución de la tolerancia al ejercicio, las infecciones pulmonares tienden a aparecer con más frecuencia, el tórax adquiere forma de barril y la falta de oxígeno confiere a los dedos forma de palillos de tambor (véase Dedos en palillo de tambor); los lechos ungueales toman un color azulado. A veces se forman pólipos en la nariz. Los senos paranasales se llenan de secreciones espesas, provocando sinusitis crónicas o recurrentes.
Los niños mayores y los adultos pueden tener episodios de estreñimiento o desarrollar obstrucción intestinal recurrente y a veces crónica. Los síntomas incluyen un cambio en el patrón de deposiciones, dolor abdominal tipo cólico, disminución del apetito y, a veces, vómitos. El reflujo gastroesofágico es relativamente frecuente entre los niños y los adultos.
Cuando un niño o un adulto con fibrosis quística suda excesivamente en un clima cálido o debido a la fiebre, puede deshidratarse por la intensa pérdida de agua y electrólitos. Los padres pueden notar la formación de cristales de sal en la piel del niño que, incluso, tiene sabor salado.
Los adolescentes presentan con frecuencia retraso del crecimiento y pubertad tardía.
A medida que la enfermedad avanza, la infección pulmonar se convierte en el mayor problema. Las infecciones pulmonares recurrentes destruyen gradualmente los pulmones.
Complicaciones de la fibrosis quística
Existen muchas complicaciones de la fibrosis quística.
La absorción insuficiente de las vitaminas liposolubles (A, D, E y K) puede causar ceguera nocturna, osteopenia (disminución en la densidad ósea), osteoporosis, anemia y trastornos hemorrágicos. En los niños que no reciben tratamiento, la mucosa interna del recto puede sobresalir por el ano, un trastorno denominado prolapso rectal. Con poca frecuencia, los niños con fibrosis quística alimentados con leches de fórmula a base de soja o hipoalergénicas desarrollan anemia y edema de las extremidades, debido a que no están absorbiendo suficientes proteínas.
Las complicaciones en adolescentes y adultos con fibrosis quística incluyen la rotura de los pequeños sacos de aire de los pulmones (alvéolos) en la cavidad pleural (el espacio entre el pulmón y la pared torácica). Esta rotura permite la entrada de aire en el espacio pleural (neumotórax), provocando el colapso del pulmón. Otras complicaciones son la insuficiencia cardíaca y la hemorragia masiva o recurrente de las vías respiratorias.
Alrededor del 2% de los niños, el 20% de los adolescentes y hasta el 50% de los adultos con fibrosis quística desarrollan diabetes, para lo cual deben tomar insulina porque el páncreas cicatrizado ya no puede producir suficiente insulina.
La obstrucción de las vías biliares por secreciones espesas causa inflamación del hígado y, finalmente, fibrosis hepática (cirrosis) en cerca del 3 - 4% de las personas con fibrosis quística. Esta cirrosis provoca aumento de la presión en las venas que llegan al hígado (hipertensión portal), dando lugar a dilatación y fragilidad de las venas del extremo inferior del esófago (varices esofágicas), que pueden romperse con facilidad y producir hemorragias importantes.
En la gran mayoría de las personas con fibrosis quística, la vesícula biliar es pequeña y se llena de una bilis espesa que no permite un correcto funcionamiento. Aproximadamente en el 10% de las personas se forman cálculos biliares, pero solo un pequeño porcentaje desarrolla síntomas. La extirpación quirúrgica de la vesícula biliar casi nunca es necesaria.
Los individuos con fibrosis quística suelen tener problemas de fertilidad. Casi todos presentan ausencia o disminución de la cantidad de espermatozoides (que los hace estériles) porque unos conductos de los testículos (los conductos deferentes) se han desarrollado de manera anómala y bloquean el paso del esperma. En las mujeres, las secreciones del cuello uterino son demasiado viscosas, lo que provoca una disminución de la fertilidad. Sin embargo, muchas mujeres con fibrosis quística han llevado a término su embarazo. El resultado del embarazo tanto para la madre como para el recién nacido está relacionado con el estado de salud de la madre durante el embarazo. Por lo demás, la funcionalidad sexual no se ve afectada ni en hombres ni en mujeres.
Otras complicaciones pueden incluir artritis, dolor crónico, problemas para dormir y apnea obstructiva del sueño, cálculos renales, enfermedad renal, depresión y ansiedad, pérdida de audición neurosensorial y zumbido de oídos (tinnitus) causados por la exposición a fármacos que dañan los oídos (especialmente los aminoglucósidos), infecciones crónicas de los senos paranasales además de un mayor riesgo de cáncer de vías biliares, páncreas e intestinos.
Diagnóstico de la fibrosis quística
Pruebas de cribado del recién nacido
Prueba del sudor
Prueba genética
Detección de portadores
Otros estudios
Evaluación del recién nacido
En Estados Unidos, las pruebas de cribado neonatal para la fibrosis quística se realizan a todos los recién nacidos. Se recoge una gota de sangre en un trozo de papel de filtro y se mide la concentración de tripsina (una enzima digestiva del páncreas). Si la concentración de tripsina en sangre es elevada, los recién nacidos se someten a pruebas confirmatorias, incluyendo pruebas de sudor y/o pruebas genéticas. La mayoría de los casos de fibrosis quística se identifican en la actualidad por medio de pruebas de cribado neonatal.
Si no se realizan pruebas de cribado neonatal, el diagnóstico de la fibrosis quística suele confirmarse al nacer o en la primera infancia, aunque a veces permanece indetectable hasta la adolescencia o los primeros años de vida adulta, lo que ocurre aproximadamente en un 10% de los casos.
Prueba del sudor
La prueba del sudor se hace a los recién nacidos con un resultado positivo del cribado y a los lactantes, niños y personas de edad avanzada que presentan síntomas que sugieren fibrosis quística. Esta prueba, que se realiza de forma ambulatoria, mide la cantidad de sal en el sudor. Para estimular la sudoración se coloca un fármaco (pilocarpina) sobre la piel y se recoge el sudor en un tubo delgado de plástico o se absorbe con un papel de filtro. A continuación, se mide la concentración de sal en el sudor. Una concentración de sal por encima de los valores normales confirma el diagnóstico en personas que presentan los síntomas de la fibrosis quística o que tienen familiares que padecen esta enfermedad. Aunque los resultados de esta prueba son válidos en recién nacidos a partir de las 48 horas de vida, hay que tener en cuenta que recoger una muestra de sudor lo suficientemente abundante en un recién nacido de menos de 2 semanas de edad aproximadamente resulta complicado.
Prueba genética
El estudio genético para el gen CFTR anómalo puede ayudar a diagnosticar la fibrosis quística en un recién nacido que presente un resultado positivo en la prueba de cribado neonatal, una persona que presente uno o más síntomas característicos o una persona que tenga un hermano o hermana con fibrosis quística. El hallazgo de 2 genes anómalos de fibrosis quística (variantes) concuerda con el diagnóstico de fibrosis quística. Sin embargo, para confirmar el diagnóstico se necesita todavía un resultado positivo de la prueba del sudor. Además, dado que los estudios genéticos típicos no detectan todas las alteraciones de las más de 2000 clases de variantes de fibrosis quística, el hecho de que no detecten 2 variantes no garantiza que la persona no tenga la fibrosis quística (aunque la posibilidad de tenerla es muy baja). La enfermedad se diagnostica mediante la realización de estudios genéticos prenatales en el feto, por medio de una biopsia de vellosidades coriónicas o mediante la amniocentesis.
Algunos bebés con un resultado positivo de la prueba para la detección neonatal de fibrosis quística pueden ser difíciles de clasificar, incluso después de la realización de pruebas del sudor y pruebas genéticas. Son bebés que no presentan síntomas relacionados con la fibrosis quística, obtienen resultados de la prueba del sudor que están entre los rangos positivos y negativos y presentan una única variante del gen de la fibrosis quística o ninguna. En este caso, los médicos establecen el diagnóstico de síndrome metabólico relacionado con RCFQ, también llamado cribado positivo de la fibrosis quística con diagnóstico no conclusivo. Aunque la mayoría de estos lactantes se mantienen sanos, en etapas posteriores de la vida alrededor del 10% desarrollan síntomas relacionados con la fibrosis quística y se les diagnostica fibrosis quística o un trastorno relacionado con la fibrosis quística. Por lo tanto, todos estos niños deben ser controlados regularmente en un centro de atención de fibrosis quística.
Algunas personas, generalmente adultos, desarrollan síntomas que afectan solo un órgano, a menudo con un resultado de prueba de sudor intermedio y sin dos variantes que causan fibrosis quística. Por ejemplo, los síntomas pueden afectar solo el páncreas (lo que da lugar a pancreatitis), los pulmones (lo que da lugar a bronquiectasia) o los órganos reproductores masculinos (dando lugar a infertilidad). El diagnóstico en estos casos es un trastorno relacionado con la conductancia transmembrana de la fibrosis quística (RCFQ).
Detección de portadores
Se puede realizar la prueba de portador en aquellas personas que deseen tener hijos o busquen consejo prenatal. En particular, los familiares de una persona con fibrosis quística suelen querer conocer cuál es la probabilidad de que tengan hijos con la enfermedad, y se les debe ofrecer la posibilidad de realizar estudios genéticos y de recibir asesoramiento. Una muestra de sangre es todo lo que se necesita para determinar si una persona tiene el gen anómalo (variante) que produce la fibrosis quística.
A no ser que los dos progenitores potenciales tengan por lo menos uno de tales variantes, sus hijos no tendrán fibrosis quística. Si ambos progenitores son portadores del gen defectuoso de la fibrosis quística, existe un 25% de probabilidades en cada embarazo de que el niño presente la enfermedad, un 50% de que tengan un hijo portador de la misma y un 25% de que tengan un hijo sin genes defectuosos para la fibrosis quística.
Otros estudios
Debido a que la fibrosis quística puede afectar varios órganos, resulta útil realizar pruebas complementarias. Los valores de las enzimas pancreáticas suelen estar por debajo de sus valores normales y el análisis de las heces revela la disminución o la carencia de la enzima digestiva elastasa, (secretadas por el páncreas) y una alta concentración de sustancias grasas.
Se hacen análisis de sangre para determinar si la concentración de insulina es inferior a la esperada y si los niveles de azúcar en sangre son elevados. También se realizan análisis de sangre para detectar problemas hepáticos y medir las concentraciones de vitaminas liposolubles.
Los médicos toman rutinariamente muestras de material de la garganta o de los esputos procedentes de los pulmones y las cultivan para identificar bacterias en las vías respiratorias, de manera que puedan decidir qué antibióticos pueden ser necesarios.
Las pruebas funcionales respiratorias pueden mostrar un trastorno respiratorio y son buenos indicadores del correcto funcionamiento de los pulmones. Estas pruebas se realizan varias veces al año y siempre que haya un deterioro de la salud de una persona.
Las radiografías de tórax y la tomografía computarizada (TC) torácica son útiles para documentar una infección pulmonar y la extensión de la lesión. La TC de los senos paranasales se realiza a aquellas personas que presentan síntomas de sinusitis grave, sobre todo si tienen pólipos nasales o se está considerando la posibilidad de someterles a una cirugía de senos paranasales.
Tratamiento de la fibrosis quística
inmunizaciones de rutina (vacunación)
Antibióticos, fármacos inhalados para diluir las secreciones de las vías respiratorias y técnicas de desobstrucción de la vía aérea para eliminar las secreciones
Fármacos que ayudan a evitar que las vías respiratorias se estrechen (broncodilatadores) y, a veces, corticoesteroides
Complementos de enzimas pancreáticas y vitaminas
Dieta alta en calorías
En personas con variantes específicas, moduladores del RCFQ
Una persona con fibrosis quística debe seguir un programa terapéutico completo dirigido por un médico especialista en fibrosis quística, generalmente un pediatra o un internista, junto con un equipo formado por otros médicos, personal de enfermería, nutricionistas, terapeutas de las respiración, e idealmente, trabajadores sociales, genetistas, farmacéuticos y profesionales de la salud mental. Los objetivos de la terapia consisten en la prevención y el tratamiento a largo plazo de los trastornos pulmonares, digestivos y demás complicaciones, así como el mantenimiento de una buena nutrición y la promoción de la actividad física.
Los niños con fibrosis quística necesitan apoyo social y psicológico, puesto que pueden ser incapaces de participar en las actividades propias de la infancia, lo que los hace sentirse aislados. La mayor parte de la carga en el tratamiento de los niños con fibrosis quística recae sobre los padres, que deben recibir información, entrenamiento y apoyo suficiente para comprender la afección y las razones de los tratamientos.
Los adolescentes necesitan orientación y formación a medida que se independizan y asumen la responsabilidad de su cuidado.
Los adultos necesitan apoyo cuando se ocupan de cuestiones relacionadas con el empleo, las relaciones, el seguro de salud y el deterioro de la salud.
Tratamientos para los pulmones
El tratamiento de los trastornos pulmonares se centra en
Prevención del bloqueo (la obstrucción) de las vías respiratorias
Controlar la infección
La persona afectada debe recibir todas las inmunizaciones rutinarias (vacunación), especialmente para las infecciones que pueden causar problemas respiratorios, como Haemophilus influenzae, gripe, sarampión, tosferina, neumococo, varicela y COVID-19.
Los tratamientos de desobstrucción de las vías respiratorias, que consisten en drenaje postural, percusión torácica, palmadas sobre la pared torácica y estimulación de la tos (véase Rehabilitación respiratoria), se inician tan pronto como se establece el diagnóstico de fibrosis quística. Los padres de un niño pequeño con la enfermedad pueden aprender estos métodos y practicarlos en casa cada día. Los niños mayores y los adultos pueden realizar estos tratamientos de manera independiente, utilizando respiradores especiales, un chaleco inflable que vibra a una frecuencia elevada (chaleco de oscilación a alta frecuencia) o maniobras de respiración especiales. El ejercicio aeróbico, practicado regularmente, también puede ayudar a mantener las vías respiratorias despejadas.
Los broncodilatadores son fármacos que ayudan a evitar el estrechamiento de las vías respiratorias. Los broncodilatadores se suelen administrar inhalados. Las personas con enfermedades pulmonares graves y una baja concentración de oxígeno en sangre requieren tratamiento con oxígeno (oxigenoterapia). La mayoría de individuos que padecen insuficiencia respiratoria crónica no obtienen ningún beneficio con el uso de un respirador artificial. Sin embargo, la respiración artificial durante periodos ocasionales y breves es beneficiosa cuando están hospitalizados por una infección aguda, después de una intervención quirúrgica o cuando esperan un trasplante de pulmón.
Los fármacos que ayudan a diluir el moco espeso de las vías respiratorias, como la dornasa alfa o la solución salina hipertónica (una solución salina altamente concentrada), se utilizan de forma generalizada. Estos fármacos se inhalan a través de un nebulizador. Estos facilitan la expectoración del esputo, mejoran la funcionalidad pulmonar y disminuyen también la frecuencia de infecciones respiratorias graves.
La administración por vía oral de corticoesteroides, como prednisona o dexametasona, puede aliviar los síntomas en lactantes con inflamación bronquial grave, en personas que sufren un estrechamiento de las vías respiratorias que no permite abrirlas con broncodilatadores y en personas que sufren una reacción alérgica pulmonar a un tipo de hongo (aspergilosis broncopulmonar alérgica). La aspergilosis broncopulmonar alérgica también se trata con un antifúngico administrado por vía oral, por vía intravenosa o por ambas vías.
El ibuprofeno, un medicamento antiinflamatorio no esteroideo (AINE), se utiliza a veces para retrasar el deterioro de la función pulmonar.
Antibióticos
Los antibióticos se utilizan a menudo para tratar tanto infecciones respiratorias agudas como crónicas en personas con fibrosis quística. Se recoge una muestra de esputo tosido o un hisopo de una muestra de la parte posterior de la garganta y las amígdalas y se analiza a intervalos regulares durante un periodo de salud estable y también durante periodos de aumento de los síntomas para que se pueda identificar el microorganismo infeccioso y el médico pueda elegir los medicamentos con más probabilidades de controlarlo. Las especies que se encuentran con mayor frecuencia son Staphylococcus aureus, incluyendo las cepas resistentes a la meticilina o las sensibles a la meticilina, y Pseudomonas. Se pueden administrar por vía oral diversos tipos de antibióticos para el tratamiento de las infecciones estafilocócicas. Para tratar una nueva infección por Pseudomonas, se administra durante 4 semanas tobramicina, aztreonam o colistina en forma inhalada.
Sin embargo, si la infección es grave o provoca un empeoramiento significativo de los síntomas o de la función pulmonar, puede ser necesario administrar antibióticos por vía intravenosa. Para este tratamiento, se combina el aminoglucósido tobramicina (también denominado en ocasiones amikacina) con otro antibiótico que ataca de forma específica a las Pseudomonas. Estos otros antibióticos son cefalosporinas, penicilinas, fluoroquinolonas y monobactámicos. Este tratamiento suele requerir hospitalización, pero parte del tratamiento también puede administrarse en el domicilio.
El tratamiento con la forma en aerosol de tobramicina o aztreonam en meses alternos durante un periodo prolongado de tiempo, además de la toma de manera continuada de una forma oral del antibiótico azitromicina 3 veces por semana, puede ayudar a controlar las infecciones crónicas por Pseudomonas y retrasar el deterioro de la función pulmonar.
Moduladores de RCFQ
Los moduladores de CFTR (RCFQ) son fármacos orales tomados de forma crónica que mejoran la función de la proteína defectuosa producida por variantes en el gen CFTR (RCFQ).
Existen 4 moduladores o combinaciones de RCFQ para las personas que presentan variantes específicas: ivacaftor, lumacaftor/ivacaftor, tezacaftor/ivacaftor y elexaftor/tezacaftor/ivacaftor. Estos medicamentos se pueden usar para tratar a cerca del 90% de las personas que sufren fibrosis quística. Los médicos administran estos medicamentos a las personas en función de su edad y las variantes que sufren.
El ivacaftor se puede administrar a pacientes de 1 mes o más de edad que tienen al menos 1 copia de una variante de fibrosis quística específica.
La combinación de lumacaftor e ivacaftor puede administrarse a personas a partir de 1 año de edad que portan 2 copias de la variante F508del.
La combinación de tezacaftor e ivacaftor puede administrarse a personas a partir de 6 años de edad que portan 2 copias de la variante F508del u otras variantes específicas.
La combinación de eleuscoftor, tezacaftor e ivacaftor puede administrarse a personas de 2 años o más que portan al menos 1 copia de la variante F508del o 1 copia de determinadas variantes poco frecuentes.
Los moduladores de CFTR pueden mejorar la función pulmonar, la función del páncreas y la calidad de vida; aumentar el peso; y disminuir la concentración de sal en el sudor y la frecuencia de infecciones pulmonares y hospitalizaciones. Aunque todos estos medicamentos pueden ser eficaces, solo ivacaftor y la combinación de elexaftor, tezacaftor e ivacaftor se consideran una terapia muy eficaz.
Los médicos están trabajando para desarrollar fármacos que ayuden a las personas con otras variantes que causan fibrosis quística.
Enemas y ablandadores de heces
Los recién nacidos cuyos intestinos están obstruidos pueden ser tratados con soluciones para enema pero a menudo requieren cirugía.
Los niños mayores y los adultos que sufren estreñimiento o tienen intestinos parcialmente obstruidos pueden ser tratados con ablandadores de heces, enemas y soluciones especiales administradas por vía oral o a través de un pequeño tubo de plástico flexible (sonda nasogástrica) que se pasa a través de la nariz o la boca hasta el estómago.
Dieta y suplementos
La dieta debe aportar suficientes calorías y proteínas para un crecimiento normal. Dado que la digestión y la absorción pueden ser insuficientes incluso cuando se utilizan los suplementos de enzimas pancreáticas, la mayoría de los niños necesitan consumir una cantidad de calorías entre un 30 y un 50% superior a la cantidad normalmente recomendada para asegurar un crecimiento suficiente. La proporción de grasas debe ser normal o incluso alta. Los suplementos orales ricos en calorías pueden proporcionar calorías adicionales para niños y adultos.
Las personas que no pueden absorber suficientes nutrientes de los alimentos requieren una alimentación complementaria, que se administra mediante una sonda introducida en el estómago o en el intestino delgado.
Las personas con fibrosis quística deben recibir el doble de la cantidad diaria recomendada de vitaminas liposolubles (A, D, E y K), contenidas en fórmulas especiales de fácil absorción.
Los medicamentos que estimulan el apetito pueden ser útiles (aunque muchas personas con fibrosis quística tienen buen apetito sin dichos medicamentos). Cuando hacen ejercicio, tienen fiebre o estén expuestas a un clima cálido, deben incrementar la ingestión de líquidos y sal.
Suplementos enzimáticos pancreáticos
Los pacientes con afectación pancreática deben tomar cápsulas de suplementos de enzimas pancreáticas en todas las comidas y refrigerios. En el caso de los bebés, los progenitores abren las cápsulas y mezclan el contenido con un alimento ácido, como la compota de manzana, para que el revestimiento especial del complemento de enzimas pancreáticas no se disuelva antes de llegar a los intestinos. Para algunas personas, los fármacos reductores del ácido estomacal, como un bloqueante de la histamina-2 o un inhibidor de la bomba de protones, pueden mejorar la eficacia de las enzimas pancreáticas. Las leches maternizadas que contienen proteínas y grasas de fácil digestión son útiles en niños con problemas pancreáticos graves y trastornos del crecimiento.
Insulina
A las personas con fibrosis quística que tienen diabetes se les deben poner inyecciones de insulina. Los medicamentos por vía oral para la diabetes no son adecuados.
El tratamiento de la diabetes en personas con fibrosis quística incluye, además del control de la insulina, asesoramiento nutricional, un programa de autocontrol de la diabetes y el seguimiento de las complicaciones oculares y renales. El asesoramiento nutricional específico es necesario porque las recomendaciones nutricionales estándar para las personas que presentan únicamente diabetes o únicamente fibrosis quística no son suficientes en los casos en que se presentan ambas enfermedades a la vez.
Cirugía
A veces es necesaria la intervención quirúrgica para tratar casos como: colapso pulmonar, sinusitis crónica, infección crónica grave en una zona determinada del pulmón, hemorragia vascular esofágica, enfermedad de la vesícula biliar u obstrucción intestinal. La hemorragia pulmonar masiva o recurrente se trata bloqueando la arteria responsable, mediante un procedimiento denominado embolización.
El trasplante de hígado ha tenido éxito en personas con hemorragia debida a varices esofágicas o daño hepático grave.
El trasplante pulmonar doble en casos de enfermedad pulmonar grave es cada día más frecuente y con mejor resultado, gracias a la experiencia y el perfeccionamiento de las técnicas. Para los adultos que sufren fibrosis quística, la supervivencia promedio después de un trasplante doble de pulmón es de unos 9 años.
Otros tratamientos
Se requieren fármacos para tratar la inflamación crónica de los senos paranasales (sinusitis), porque este problema es muy frecuente. Las opciones de tratamiento consisten en hacer enjuagues con una solución de agua salada introducida por la nariz (irrigación con solución salina nasal), inhalar dornasa alfa con un nebulizador e irrigar la nariz y los senos paranasales con antibióticos. Se recomienda el uso de un aerosol nasal con corticoesteroides para tratar la inflamación y la hinchazón de las membranas mucosas de la nariz (rinitis alérgica).
A las personas con insuficiencia cardíaca se les administran fármacos (diuréticos) para reducir la cantidad de líquido que retienen. Los diuréticos son eficaces porque aumentan la cantidad de agua que los riñones pueden evacuar del cuerpo. También debe limitarse el consumo de sal común y de los alimentos salados.
Las inyecciones de la hormona de crecimiento humana puede mejorar la función pulmonar, aumentar la talla y el peso, y reducir la tasa de hospitalización. Sin embargo, este fármaco es caro y supone inconvenientes para las personas que lo reciben, por lo que los médicos no suelen prescribirlo.
Algunas personas con niveles bajos de oxígeno en la sangre pueden necesitar oxígeno complementario que, normalmente, se administra a través de un tubo nasal con dos extremos (cánula). Algunas personas son tratadas mediante una máscara bien ajustada colocada sobre la nariz o la nariz y la boca. Se suministra una mezcla de oxígeno y aire a presión a través de la máscara. Esta técnica, denominada presión positiva de las vías respiratorias en dos niveles (BiPAP) o presión positiva continua en las vías respiratorias (CPAP), puede ayudar a las personas afectadas a mantener niveles normales de oxígeno mientras duermen.
Problemas del enfermo terminal
Las personas que sufren fibrosis quística y sus familiares necesitan hablar con su médico y equipo de atención sobre su pronóstico y sobre el tipo de tratamiento que desean recibir. Estas conversaciones son especialmente importantes para las personas cuya función pulmonar está empeorando. La persona afectada necesita estar preparada para lo que está por venir y saber qué tratamientos se pueden hacer para prolongar la vida. Cuando la fibrosis quística está avanzada, las personas afectadas y sus familias necesitan discutir los beneficios potenciales y los problemas de un trasplante pulmonar.
Los médicos deben dar a los afectados por fibrosis quística la información que necesitan para tomar decisiones sobre su atención y deben ayudarles a determinar cómo y cuándo aceptar la muerte y cómo hablar de ella.
Cuando los tratamientos agresivos ya no son eficaces, los médicos pueden comenzar a tratar a las personas afectadas con tratamientos que sólo apuntan a aliviar los síntomas (llamados cuidados paliativos). Es mucho mejor para la persona afectada poder tomar este tipo de decisiones respecto a la atención médica terminal mucho antes de que sea necesario. Las decisiones tomadas con antelación son muy importantes porque, si se espera demasiado, la enfermedad suele impedir al afectado manifestar su voluntad. Este proceso de toma de decisiones con anticipación se denomina voluntades anticipadas. Dicha planificación debe implicar la ejecución de documentos legales apropiados que reflejen los deseos de la persona afectada con respecto a la atención médica al final de su vida.
Pronóstico de la fibrosis quística
La gravedad de la fibrosis quística varía mucho de una persona a otra con independencia de la edad, y depende en gran medida del grado en que los pulmones resultan afectados. En Estados Unidos, se predice que las personas con fibrosis quística que nacieron en el año 2021 vivirán aproximadamente hasta los 65 años de edad. El pronóstico para una supervivencia más larga ha mejorado constantemente, principalmente porque los afectados son diagnosticados antes y en la actualidad los tratamientos pueden posponer algunas de las alteraciones que ocurren en los pulmones. La supervivencia a largo plazo es significativamente mejor en las personas que no llegan a tener problemas pancreáticos.
Sin embargo, el deterioro es inevitable, lo que lleva a la pérdida de la funcionalidad pulmonar y finalmente a la muerte. Las personas con fibrosis quística generalmente mueren de insuficiencia respiratoria después de muchos años de deterioro de la funcionalidad pulmonar. Un pequeño número, sin embargo, muere de insuficiencia cardíaca, enfermedad hepática, hemorragia en las vías respiratorias o complicaciones de la cirugía, incluyendo trasplante de pulmón y/o hígado. A pesar de sus muchos problemas, las personas con fibrosis quística pueden llevar una vida productiva y a menudo asisten a la escuela o al trabajo hasta poco antes de morir.
Más información
El siguiente recurso en inglés puede ser útil. Tenga en cuenta que LOS MANUALES no se hacen responsables del contenido de ningún otro recurso que no sea el último.
Cystic Fibrosis Foundation (Fundación para la Fibrosis Quística): un recurso que proporciona información sobre las opciones de tratamiento disponibles, el desarrollo de fármacos, la investigación y los servicios de apoyo comunitario




