




El complejo de esclerosis tuberosa es un trastorno genético de herencia dominante en el que aparecen tumores (en general, hamartomas) en múltiples órganos. El diagnóstico requiere criterios clínicos específicos y estudios de diagnóstico por imágenes del órgano afectado. El tratamiento es sintomático o, si los tumores del sistema nervioso central están creciendo, farmacoterapia con sirolimús o everolimús. Debe controlarse con regularidad a los pacientes para detectar complicaciones.
El complejo de esclerosis tuberosa es un síndrome neurocutáneo que se produce en 1 cada 6000 niños; 85% de los casos involucra mutaciones en el gen TSC1 (9q34), que controla la producción de hamartina, o el gen TSC2 (16p13.3), que controla la producción de tuberina. Estas proteínas actúan como supresores del crecimiento. Si alguno de los progenitores presenta el trastorno, los hijos tienen un riesgo del 50% de tenerlo. Sin embargo, dos tercios de los casos se deben a mutaciones nuevas.
Los pacientes con complejo de esclerosis tuberosa tienen tumores o anomalías que se manifiestan a diferentes edades y en múltiples órganos, que incluyen
Encéfalo
Corazón
Ojos
Riñones
Pulmones
Piel
Los tubérculos del sistema nervioso central interrumpen los circuitos neuronales, y producen retraso en el desarrollo y deterioro cognitivo y pueden causar convulsiones, incluidos los espasmos infantiles. A veces los tubérculos crecen y obstruyen el flujo del líquido cefalorraquídeo desde los ventrículos laterales, lo que provoca hidrocefalia unilateral. A veces, los tubérculos sufren degeneración maligna en gliomas, particularmente astrocitomas subependimarios de células gigantes (SEGA).
Se pueden desarrollar rabdomiomas cardíacos antes del nacimiento, que a veces causan insuficiencia cardíaca en los recién nacidos. Estos rabdomiomas tienden a desaparecer con el tiempo y por lo general no causan síntomas en la niñez o en la edad adulta.
Se pueden desarrollar tumores renales (angiolipomas) en los adultos, y la enfermedad poliquística del riñón pueden desarrollarse a cualquier edad. La enfermedad renal puede causar hipertensión.
Se pueden desarrollar lesiones pulmonares, como la linfangioleiomiomatosis, sobre todo en las adolescentes.
Signos y síntomas del complejo de esclerosis tuberosa
Las manifestaciones varían mucho en gravedad. Por lo general se presentan lesiones cutáneas.
Los lactantes con lesiones del sistema nervioso central pueden presentar un tipo de convulsión denominado espasmos de la lactancia. Los niños afectados también pueden tener convulsiones, discapacidad intelectual, autismo, trastornos de aprendizaje o problemas conductuales.
Los parches acrómicos en la retina, así como los hamartomas retinianos, son comunes y pueden ser visibles en el fondo de ojo.
Son frecuentes las picaduras de esmalte en los dientes permanentes.
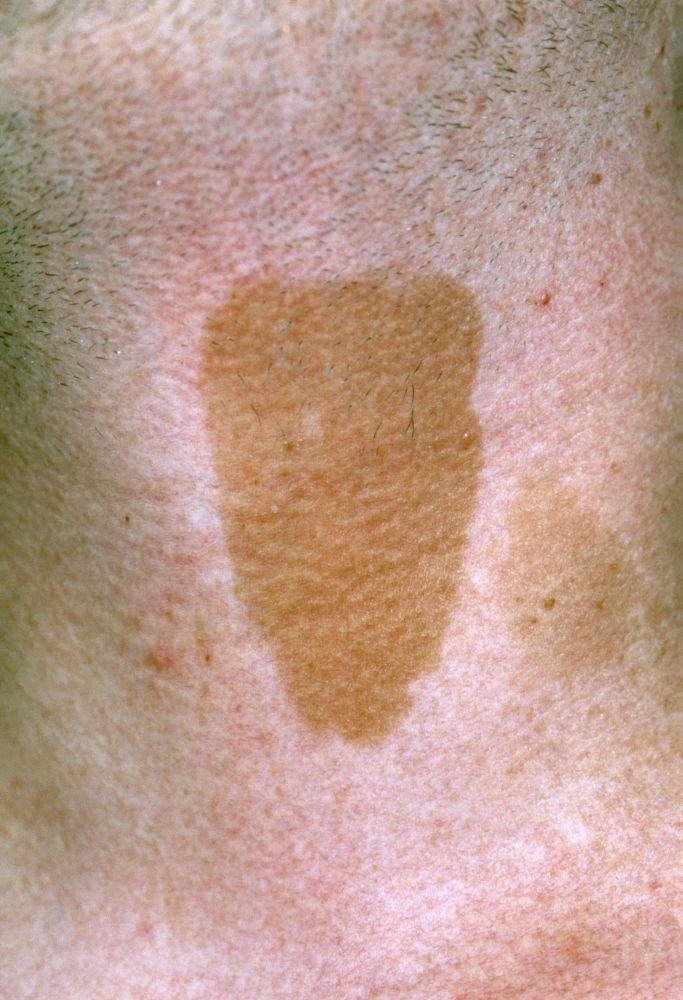
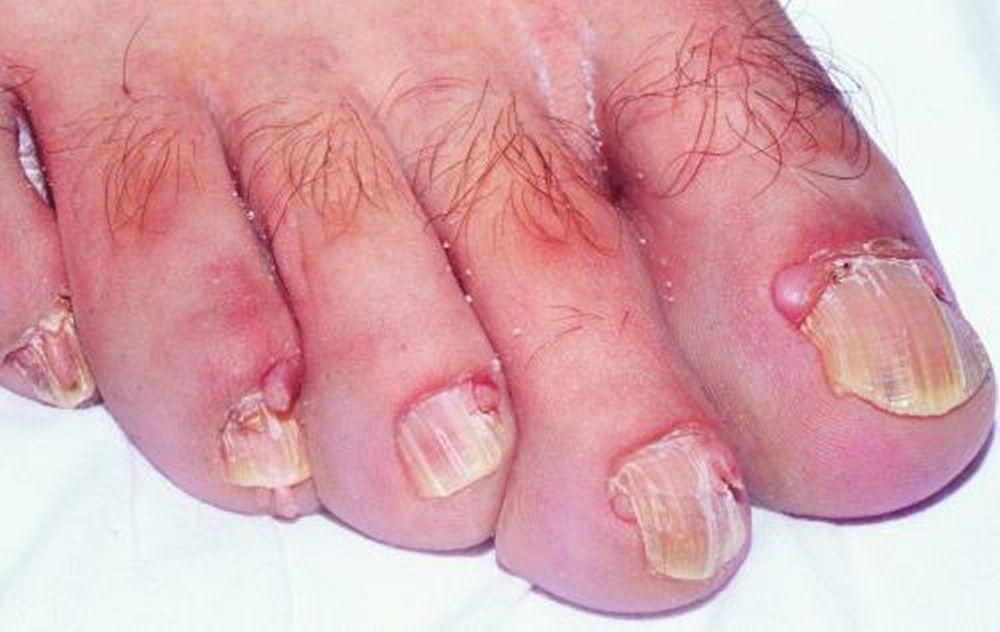
Los hallazgos cutáneos son
Máculas inicialmente pálidas en forma de hoja de fresno, que aparecen durante la lactancia o la primera infancia
Angiofibromas de la cara (adenomas sebáceos), que aparecen en etapas más tardías de la infancia
Parches de piel de zapa congénitos (lesiones sobreelevadas que se asemejan a una cáscara de naranja), por lo general en la espalda
Nódulos subcutáneos
Manchas café con leche
Fibromas subungueales, que pueden aparecer en cualquier momento durante la infancia o en etapas tempranas de la adultez

Con autorización del editor. From Puduvalli V: Atlas of Cancer. Publicado por M Markman y R Gilbert. Philadelphia, Current Medicine, 2002.
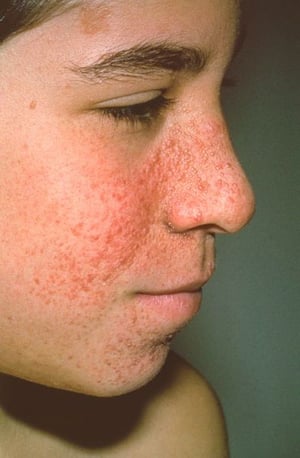
Imagen cortesía de Karen McKoy, MD.
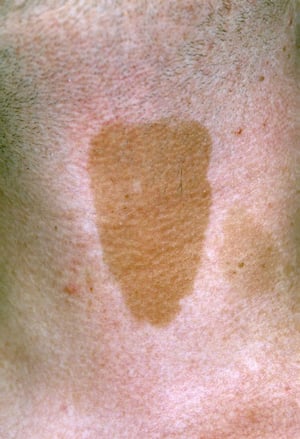
DR P. MARAZZI/SCIENCE PHOTO LIBRARY
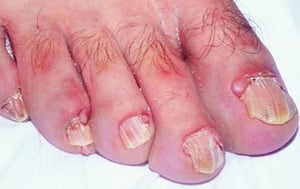
© Springer Science+Business Media


Con autorización del editor. From Puduvalli V: Atlas of Cancer. Publicado por M Markman y R Gilbert. Philadelphia, Current Medicine, 2002.

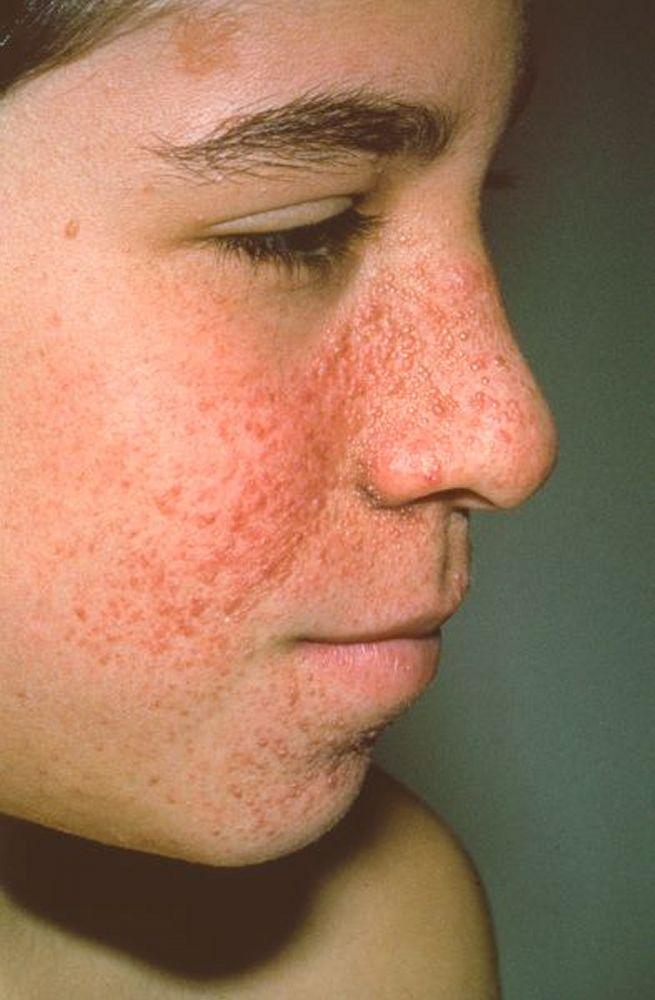
Imagen cortesía de Karen McKoy, MD.

DR P. MARAZZI/SCIENCE PHOTO LIBRARY

© Springer Science+Business Media
Diagnóstico del complejo de esclerosis tuberosa
Criterios clínicos
Identificación de las lesiones cutáneas
Estudios por imagen de los órganos afectados
Evaluación genética molecular
La Conferencia de consenso internacional sobre el complejo de esclerosis tuberosa de 2012 definió los criterios mayores y menores para realizar un diagnóstico definitivo o posible de CET (1; véase tabla Criterios de la Conferencia de Consenso Internacional para el diagnóstico del complejo de esclerosis tuberosa).
Un diagnóstico definitivo de CET con estos criterios requiere alguno de los siguientes:
Identificación de una mutación TSC1 o TSC2 patógena mediante pruebas genéticas moleculares
Dos características mayores o 1 característica mayor con ≥ 2 características menores
Un diagnóstico posible de CET con estos criterios requiere los siguientes elementos:
1 característica mayor o ≥ 2 características menores
Se realiza un examen físico para detectar lesiones cutáneas típicas. Se debe realizar un fondo de ojo para detectar parches acrómicos en la retina.
La esclerosis tuberosa complejo se puede sospechar cuando la ecografía fetal detecta rabdomiomas cardíacos o cuando se producen los espasmos infantiles.
Las manifestaciones cardíacas o craneales pueden ser visibles en la ecografía prenatal de rutina. Se requiere RM o ecografía de los órganos afectados para confirmar el diagnóstico.
Existen estudios genéticos específicos.
Referencia del diagnóstico
1. Northrup H, D Krueger D, and on behalf of the International Tuberous Sclerosis Complex Consensus Group: Tuberous sclerosis complex diagnostic criteria update: Recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol 49(4):243–254, 2013. doi: 10.1016/j.pediatrneurol.2013.08.001
Pronóstico del complejo de esclerosis tuberosa
El pronóstico depende de la gravedad de los síntomas. Los lactantes con síntomas leves suelen evolucionar bien y vivir vidas largas y productivas; los lactantes con síntomas graves pueden tener discapacidades graves.
Independientemente de la gravedad, la mayoría de los niños muestran progreso continuado del desarrollo.
Tratamiento del complejo de esclerosis tuberosa
Tratamiento sintomático
Sirolimús o everolimús
El tratamiento del complejo de esclerosis tuberosa es tanto sintomático como específico:
Para las convulsiones: anticonvulsivos (especialmente vigabatrina para los espasmos infantiles) o algunas veces cirugía de la epilepsia
Para las lesiones de piel: dermoabrasión o técnicas láser
Para problemas neuroconductuales: técnicas de intervención conductual o fármacos
Para hipertensión causada por problemas renales: antihipertensivos o cirugía para resecar tumores en crecimiento
Para retrasos del desarrolo: escolaridad especial o terapia ocupacional
Para los tumores malignos y algunos de los tumores benignos: everolimús o sirolimús
Aunque hasta el momento solo están aprobados para el tratamiento de astrocitomas subependimarios de células gigantes, la evidencia creciente sugiere que sirolimús oral y su derivado, everolimús, pueden usarse para prevenir y tratar la mayoría de las complicaciones del complejo de esclerosis tuberosa. En algunos pacientes se ha demostrado que estos medicamentos encogen los tubérculos cerebrales, los rabdomiomas cardíacos que son demasiado grandes para ser resecados y las lesiones faciales y disminuyen la incidencia de convulsiones. El sirolimús tópico puede ser útil para los angiofibromas faciales (1). En la actualidad se realizan estudios que usan estos medicamentos para las complicaciones mencionadas del complejo de esclerosis tuberosa y otras.
Está indicado asesoramiento genético en adolescentes y adultos en edad fértil.
Pruebas de cribado en busca de complicaciones
Todos los pacientes deben ser evaluados de manera regular para detectar complicaciones de esclerosis tuberosa complejo de forma temprana.
En general, se realiza una de las siguientes intervenciones:
RM de la cabeza para comprobar si hay complicaciones intracraneales por lo menos cada 3 años (p. ej., astrocitomas subependimarios de células gigantes [SEGA])
Ecografía renal o RM del abdomen para detectar tumores renales cada 3 años en niños en edad escolar y cada 1 a 2 años durante toda la vida en adultos afectados
En niñas ≥ 18 años, detección sistemática de disnea durante el ejercicio y disnea habitual cada año y TC de alta resolución cada 5 a 10 años para detectar linfangioleiomiomatosis
Pruebas neuropsicológicas periódicamente y pruebas de cribado conductuales en los niños para ayudar a planificar el apoyo en la escuela y las intervenciones conductuales
Ecocardiografía al menos cada 3 años en niños y adolescentes asintomáticos con rabdomiomas cardíacos
La monitorización regular debe continuarse hasta la involución de los rabdomiomas cardíacos.
La monitorización clínica también es importante y a veces exige pruebas más frecuentes. El desarrollo de cefaleas, pérdida de habilidades, o nuevos tipos de convulsiones puede ser causado por la degeneración maligna o el crecimiento de los tubérculos del sistema nervioso central y son indicaciones de neuroimagen.
Referencia del tratamiento
1. Darling T: Topical sirolimus to treat tuberous sclerosis complex (TSC). JAMA Dermatol 154(7):761–762, 2018. doi: 10.1001/jamadermatol.2018.0465









