

Cystinuria is a rare hereditary kidney disorder that results in excretion of the amino acid cystine into the urine, often causing cystine stones to form in the urinary tract.
Cystinuria is caused by defective genes.
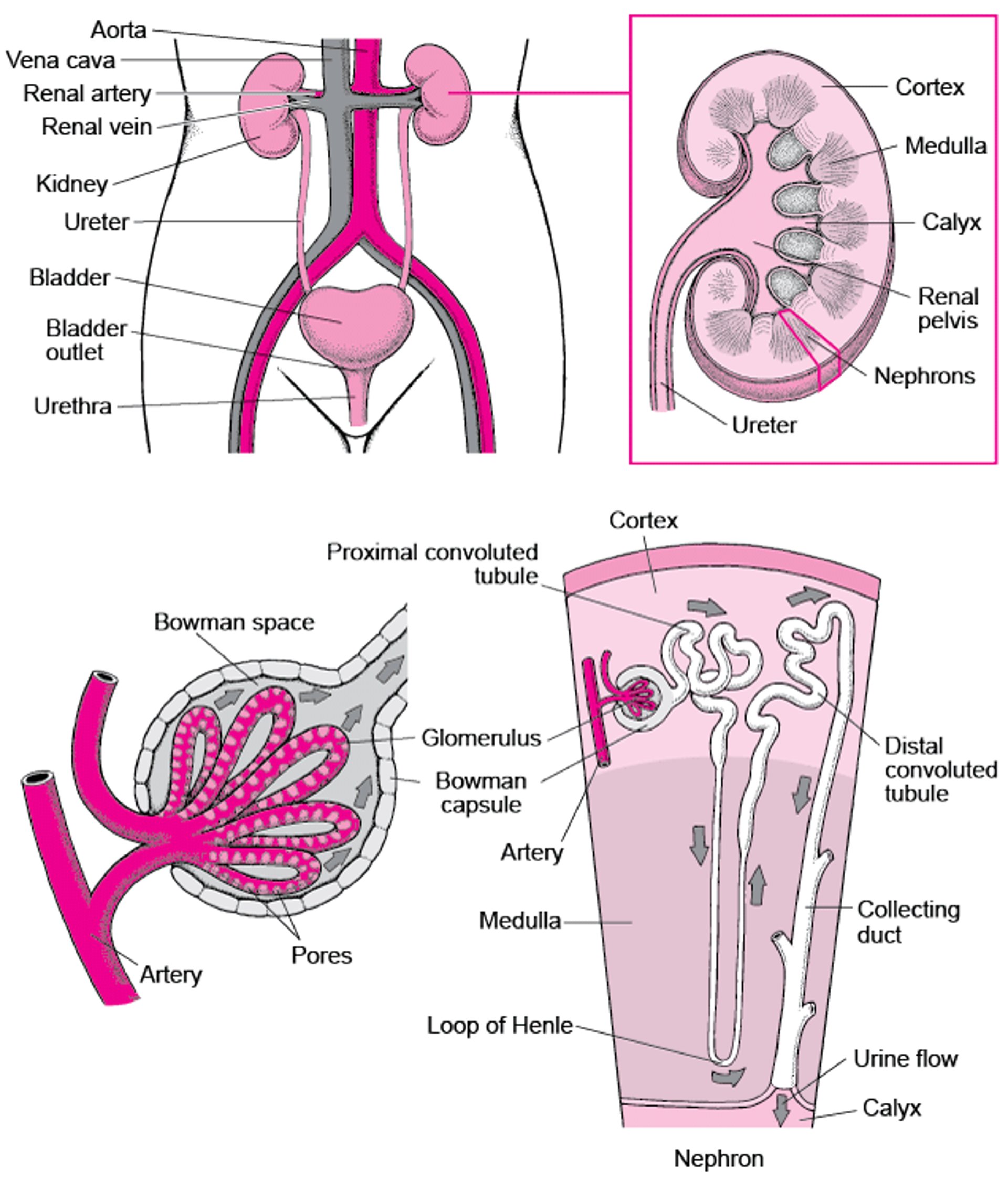
Stones form in the kidneys, bladder, renal pelvis, or ureters.
The stones are collected and analyzed, and urine tests are done.
People increase the amount the drink and may be given certain medications.
(See also Introduction to Congenital Kidney Tubular Disorders and Kidney Stones.)

Cystinuria is caused by an inherited defect of the kidney tubules. The defect causes people to excrete excessive amounts of the amino acid cystine into the urine (amino acids are the building blocks of proteins). The excess cystine causes stones to form in the kidneys, bladder, renal pelvis (the area where urine collects and flows out of the kidney), or ureters (the long, narrow tubes that carry urine from the kidneys to the bladder).
There are 2 gene abnormalities that cause most cystinuria. The genes are recessive, so people with the disorder must have inherited 2 abnormal genes, 1 from each parent (see figure ). Because 2 genes are needed when a recessive gene is involved, the parents are carriers of the gene but do not have the syndrome. However, siblings of children with the disorder might have it.
Sometimes, a person who has only one abnormal gene (a carrier), such as a parent of a person who has cystinuria, may excrete larger than normal amounts of cystine into the urine but seldom enough to form cystine stones.
Boys are generally affected more than girls.
Viewing the Urinary Tract

Symptoms of Cystinuria
Although symptoms of cystinuria may occur in infants, they usually start between the ages of 10 years and 30 years.
Often, the first symptom is intense pain caused by a spasm of the ureter where a stone becomes lodged. The stone may also become a site where bacteria collect and cause a urinary tract infection or, occasionally, kidney failure.
Diagnosis of Cystinuria
Analysis of kidney stones
Urine tests
A doctor tests for cystinuria when a person has recurring kidney stones. Stones that have been collected are analyzed.
Cystine crystals may be seen during a microscopic examination of the urine (urinalysis), and high cystine levels are found in the urine.
Treatment of Cystinuria
Increasing fluid intake
Medications to make the urine more alkaline
Medications to dissolve cystine
Decreasing salt, animal protein, and methionine in the diet
Treatment of cystinuria consists of preventing cystine stones from forming by keeping the concentration of cystine in the urine low. To keep the cystine concentration low, a person must drink enough fluids to produce at least 4 to 8½ pints (2 to 4 liters) of urine each day. During the night, however, when the person is not drinking, less urine is produced and stone formation is more likely. This risk is reduced by drinking fluids before going to bed.
Another treatment approach involves taking potassium citrate or potassium bicarbonate to make the urine more alkaline (that is, less acidic) because cystine dissolves more easily in alkaline urine than in acidic urine. Efforts to increase intake of water and make the urine more alkaline can lead to abdominal bloating, making the treatment difficult for some people to tolerate. Another treatment approach involves taking potassium citrate or potassium bicarbonate to make the urine more alkaline (that is, less acidic) because cystine dissolves more easily in alkaline urine than in acidic urine. Efforts to increase intake of water and make the urine more alkaline can lead to abdominal bloating, making the treatment difficult for some people to tolerate.
Consuming less salt, animal protein, and methionine (found in many animal proteins, cheeses, dairy, beans, and nuts) may help reduce the concentration of cystine in the urine. However, because protein is essential for proper growth, caregivers should work with doctors to make sure children sufficient amounts of it.
If stones continue to form despite these measures, medications such as penicillamine and tiopronin may be tried. These medications react with cystine to keep it dissolved. If stones continue to form despite these measures, medications such as penicillamine and tiopronin may be tried. These medications react with cystine to keep it dissolved.
Penicillamine is effective in keeping the concentration of cystine in the urine low but it can cause toxic side effects, such as fever, rash, and joint pain. Doctors give Penicillamine is effective in keeping the concentration of cystine in the urine low but it can cause toxic side effects, such as fever, rash, and joint pain. Doctors givevitamin B6 (pyridoxine) supplements to help limit the side effects. (pyridoxine) supplements to help limit the side effects.
Although the medications are usually effective, there is a fairly high risk that stones will continue to form.





