


Ohren können bei der Geburt fehlen, verformt oder unvollständig entwickelt sein.
(Siehe auch Einführung in angeborene kraniofaziale und muskuloskelettale Störungen und Überblick über angeborene kraniofaziale Anomalien.)
Eine Mikrotie und eine Atresie des Gehörgangs (die eine Schallleitungsschwerhörigkeit verursacht) betreffen das äußere Ohr. Diese Fehlbildungen, die häufig nebeneinander vorkommen, werden oft bei oder nach der Geburt identifiziert. Manchmal wird auch erst beim Schuleingangstest ein partiell verschlossener äußerer Gehörgang bei einer normalen Ohrmuschel entdeckt.
Tiefliegende Ohren sind Ohren, die unterhalb der Stelle am Kopf, wo Ohren typischerweise sind, positioniert sind. Bei tief angesetzten Ohren befindet sich die Spitze der Ohrmuschel unterhalb der horizontalen Linie, die die äußeren Augenwinkel verbindet. Diese Anomalie ist mit einer Reihe von genetischen Syndromen verbunden und oft mit Entwicklungsverzögerungen.
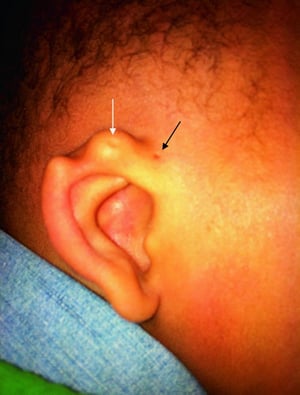
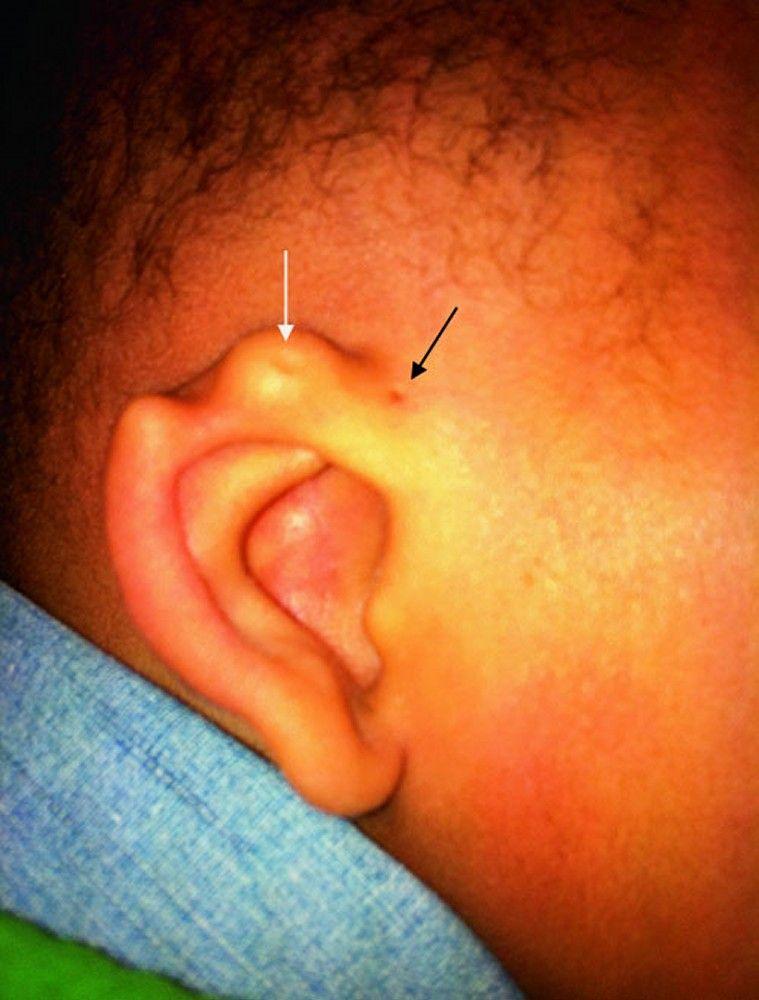
Ohrlöcher und Ohrmarken sind kleine Anomalien, die typischerweise vor dem Ohr liegen. Patienten mit diesen Anomalien sollten auf Hörverlust und auf andere angeborene Anomalien untersucht werden (z. B. Nierenanomalien mit Ohrmuscheln bei Branchio-oto-renalem Syndrom). Die Häufigkeit von Nierenanomalien ist bei Menschen mit Ohrmuscheln erhöht, sodass eine Ultraschalluntersuchung der Nieren in Betracht gezogen werden sollte.
Hörtests und eine CT des Os temporale sind notwendig, um mögliche zusätzliche Knochenfehlbildungen auszuschließen.
© Springer Science+Business Media
© Springer Science+Business Media
© Springer Science+Business Media
© Springer Science+Business Media

© Springer Science+Business Media

© Springer Science+Business Media

© Springer Science+Business Media

© Springer Science+Business Media
Diagnose
Klinisches Erscheinungsbild
Gentests
Ein klinischer Genetiker sollte betroffene Patienten auch in Fällen von offensichtlich isolierten kongenitalen Anomalien untersuchen.
Bei der Untersuchung von Patienten mit kraniofazialen Anomalien sollten eine Chromosomen-Mikroarray-Analyse, spezifische Gentests oder umfassendere Gen-Panel-Tests in Betracht gezogen werden. Wenn die Ergebnisse dieser Tests nicht diagnostisch sind, wird eine vollständige Exom-Sequenzanalyse empfohlen.
Behandlung von angeborenen Ohranomalien
Operative Eingriffe
Hörgerät
Die Behandlung von Ohrannomalien kann eine Operation und ein Hörgerät einschließen, je nachdem, ob die Fehlbildung unilateral oder bilateral vorliegt, ob sie das Hören, Lernen oder den sozialen Umgang beeinträchtigt und ob Komplikationen (z. B. Beteiligung des Fazialisnervs, Cholesteatom, Otitis media) vorhanden sind. Der chirurgische Eingriff kann aus einer Rekonstruktion der Ohrmuschel und der Schaffung eines äußeren Gehörgangs, Trommelfells und Gehörknöchelchen bestehen.