


Die gemischte Bindegewebekrankheit ist ein eher seltenes, spezifisch definiertes, Syndrom, das die klinischen Merkmale eines systemischen Lupus erythematodes, einer systemischen Sklerose sowie einer Polymyositis vereint und darüber hinaus durch den Nachweis von hochtitrigen antinukleären Antikörpern gegen Ribonukleoprotein gekennzeichnet ist. Handschwellungen, Raynaud-Syndrom, Polyarthralgie, entzündliche Myopathie, Ösophagus-Hypomotilität und interstitielle Lungenerkrankung sind häufig. Die Diagnose wird durch die Kombination typischer klinischer Merkmale, den Nachweis von Antikörpern gegen Ribonukleoprotein und das Fehlen von Autoantikörpern, die typisch sind für eine andere Systemkrankheit, gestellt. Die Behandlung hängt von der Schwere der Erkrankung und Organbeteiligung ab, umfasst aber in der Regel Kortikosteroide und zusätzlich Immunsuppressiva.
Gemischte Bindegewebserkrankungen treten weltweit und in allen Rassen auf, wobei die Häufigkeit in der Adoleszenz und in den 20er Jahren am höchsten ist. Über 80% der Menschen, die diese Krankheit haben, sind Frauen. Die Ursache von MCTD ist unbekannt. bei vielen Patienten entwickelt sich die Erkrankung zur klassischen systemischen Sklerose oder systemischen Lupus erythematodes.
Symptome und Anzeichen von MCTD
Ein Raynaud-Syndrom (Raynaud-Syndrom) kann anderen Manifestationen um Jahre vorausgehen. Häufig ähneln die ersten Manifestationen den frühen Formen eines systemischen Lupus erythematodes, einer systemischen Sklerose, einer Polymyositis oder auch einer rheumatoiden Arthritis. Viele Patienten scheinen zu Beginn eine undifferenzierte Bindegewebserkrankung zu haben. Die Erkrankung tendiert zum Fortschreiten und zur Ausbreitung, das klinische Bild ändert sich oft im Verlauf.
Die anfängliche, diffuse Schwellung der Hände ist typisch, aber nicht universell. An der Haut sieht man Ausschläge, die an einen SLE oder eine Dermatomyositis erinnern. Diffuse systemische skleroseartige Hautveränderungen und ischämische Nekrosen oder Ulzerationen der Fingerspitzen können auftreten.

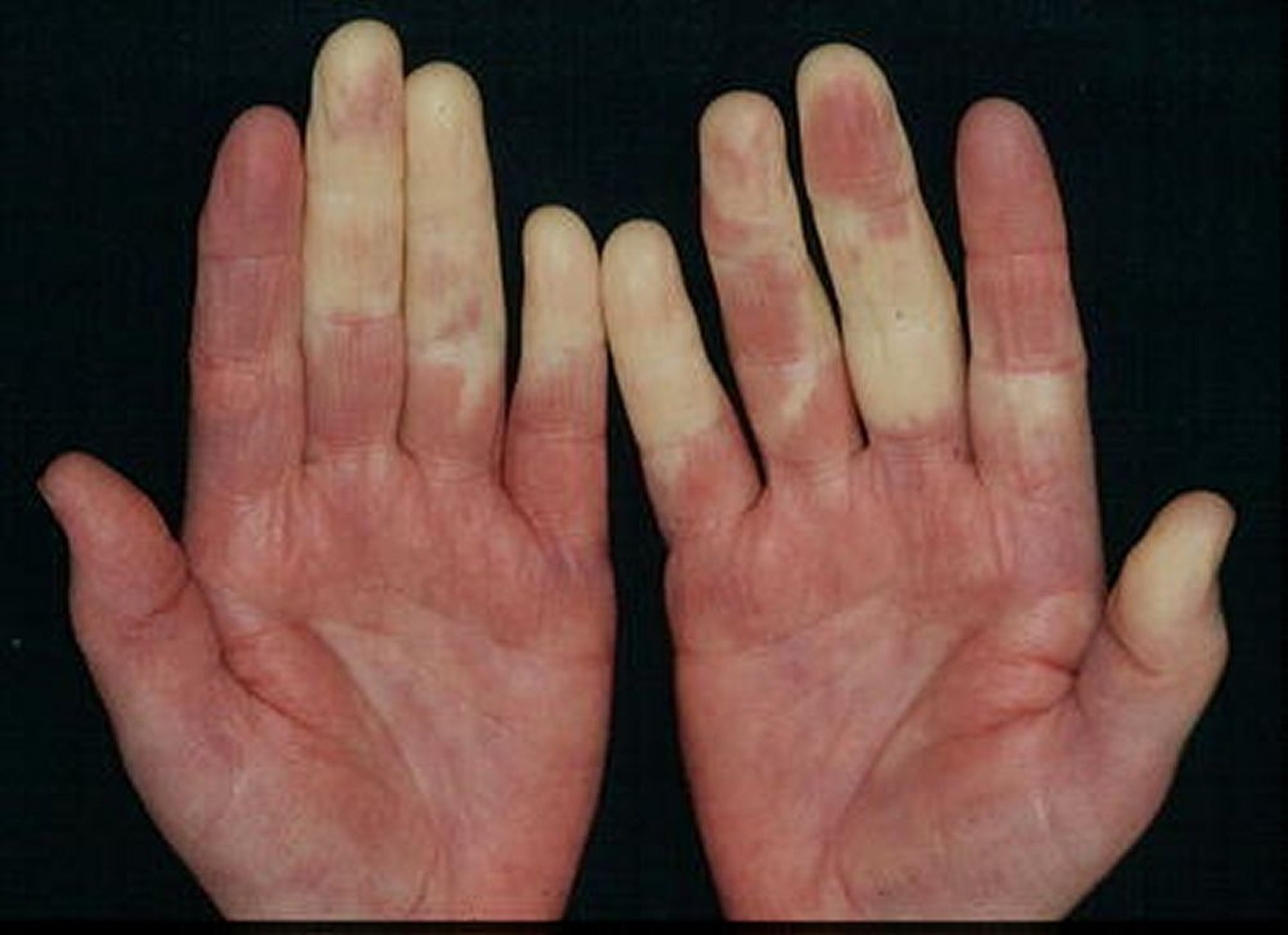
© Springer Science+Business Media

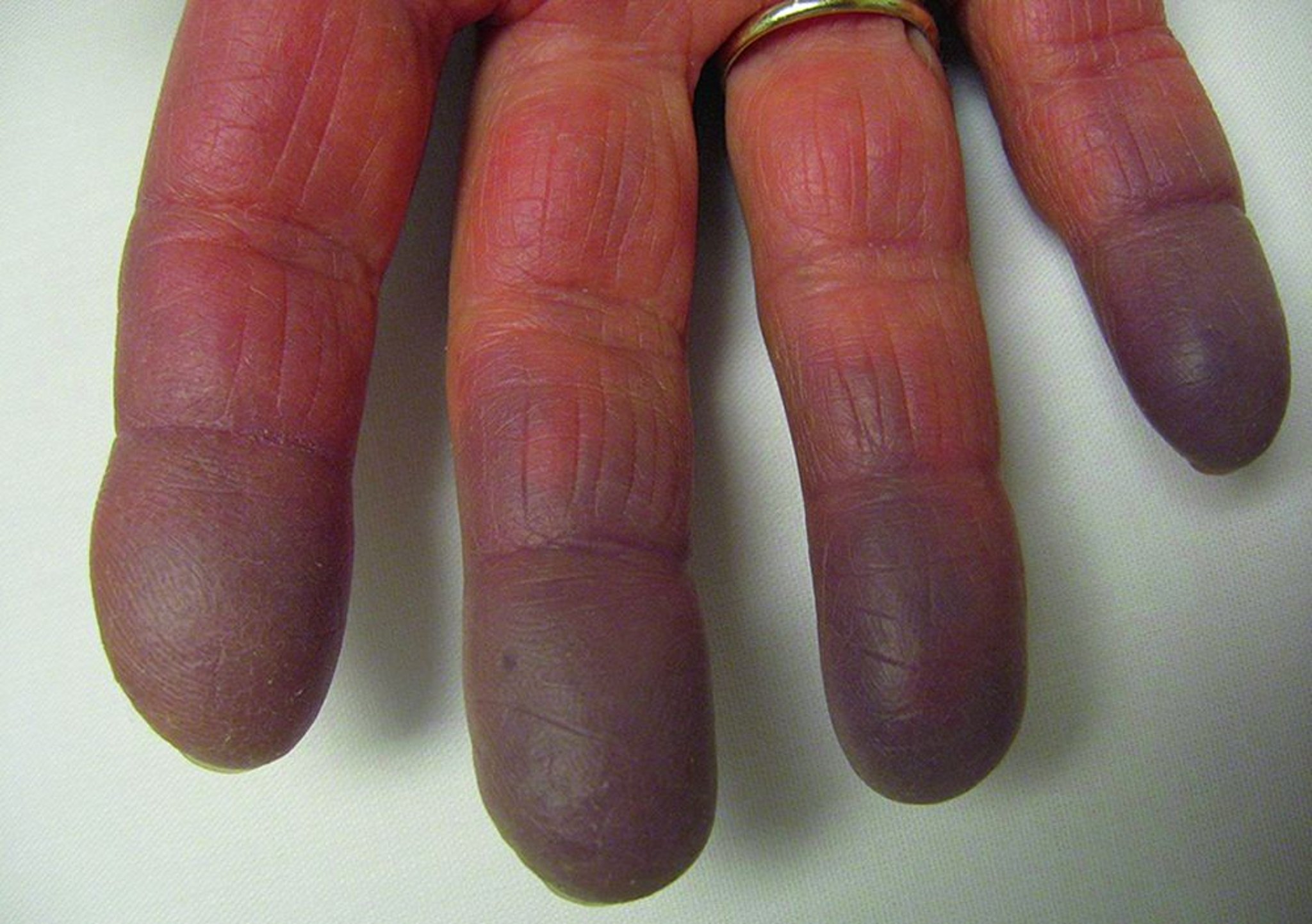
© Springer Science+Business Media
Nahezu alle Patienten weisen Polyarthralgien auf, 75% haben eine eindeutige Arthritis. Meist handelt es sich um eine nichterosive Form, es können jedoch auch erosive Veränderungen und Deformitäten ähnlich wie bei der rheumatoiden Arthritis (z. B. Knopfloch- und Schwanenhalsdeformitäten) auftreten. Proximale Muskelschwäche mit oder ohne Schmerzhaftigkeit ist häufig, gewöhnlich unter Leuten, die erhöhte Niveaus der Muskelenzyme (z. B. Creatin-Kinase) haben.
Eine renale Beteiligung (am häufigsten membranöse Nephropathie) tritt bei etwa 25% der Patienten auf und ist in der Regel mild; eine schwere Beteiligung mit Morbidität oder Mortalität ist untypisch für MCTD. Die Lungen sind bei bis zu 75% der Patienten mit MCTD betroffen. Häufigste Manifestation der Lunge ist die interstitielle Lungenerkrankung; die pulmonale Hypertonie ist eine der häufigsten Todesursachen. Es kann eine Herzinsuffizienz auftreten. Ein Sjögren-Syndrom kann sich entwickeln.
Diagnose von MCTD
Untersuchung auf antinukleäre Antikörpern, Antikörper gegen extrahierbare nukleäre Antigene (Antikörper gegen U1-Ribonukleoprotein oder RNP) und Smith- (SM) und Anti-DNA-Antikörper
Bestimmung der Organbeteiligung wie klinisch indiziert
An eine MCTD sollte gedacht werden, wenn bei Patienten mit vermeintlichem systemischen Lupus erythematodes,systemischer Sklerose oder Polymyositis zusätzliche überlappende Symptome auftreten.
Tests auf antinukleären Antikörpern und Antikörper gegen U1-RNP-Antigen werden zuerst durchgeführt. Fast alle Patienten haben hohe Titer fluoreszierender antinukleärer Antikörper, die ein gesprenkeltes Muster erzeugen. Antikörper gegen U1 RNP sind vorhanden, meist in sehr hohen Titern. Antikörper gegen die ribonukleaseresistente Sm-Komponente von extrahierbarem nukleärem Antigen (Anti-Sm-Antikörper) und gegen doppelsträngige DNA (negativ bei MCTD per Definition) werden gemessen, um andere Erkrankungen auszuschließen. Das Vorhandensein von Anti-RNP-Antikörpern reicht nicht aus, um die Diagnose einer MCTD zu stellen; es sind auch typische klinische Befunde erforderlich.
Der RF ist oft vorhanden und hochtitrig, Die Erythrozytensedimentationsrate häufig erhöht.
Eine pulmonale Hypertonie sollte so früh wie möglich mit Lungenfunktionsprüfung und Echokardiographie erkannt werden. Die weitere Abklärung hängt vom vorhandenen Spektrum an Symptomen und Befunden ab, entsprechend gestaltet sich die Suche bei Hinweisen auf eine Myositis sowie auf renale oder pulmonale Beteiligung.
Kreatinkinase, MRT, Elektromyogramm oder Muskelbiopsie können helfen, das Vorhandensein von Myositis im Rahmen von MCTD zu bestätigen.
Prognose der MCTD
Die generelle 10-Jahres-Überlebensrate liegt bei ca. 80%, im Einzelfall wird die Prognose durch die vorhandenen Manifestationen bestimmt. Patienten mit Merkmalen der systemischen Sklerose und Polymyositis haben eine schlechtere Prognose. Patienten haben ein erhöhtes Risiko für Atherosklerose. Als mögliche Todesursachen kommen pulmonale Hypertonie, Nierenversagen, Myokardinfarkt, Kolonperforation, Sepsis oder Hirnblutung in Frage. Manche Patienten haben jedoch über viele Jahre anhaltende Remissionen ohne Therapiebedarf.
Behandlung von MCTD
Nichtsteroidale entzündungshemmende Medikamente (NSAR) oder Antimalariamittel (z. B. Hydroxychloroquin, Chloroquin) bei leichter Erkrankung
Kortikosteroide und andere Immunsuppressiva (z. B. Methotrexat, Azathioprin, Mycophenolatmofetil) bei mittelschwerer bis schwerer Erkrankung
Kalziumkanalblocker (z. B. Nifedipin) und manchmal Phosphodiesteraseinhibitoren (z. B. Tadalafil) für das Raynaud-Syndrom
Das allgemeine Management und die initiale medikamentöse Therapie sind auf das spezifische klinische Problem zugeschnitten und sind vergleichbar mit denen bei systemischem Lupus erythematodes oder dem dominierenden klinischen Phänotyp. Die meisten Patienten mit mittelschwerer oder schwerer Erkrankung reagieren auf Kortikosteroide, besonders wenn sie früh behandelt werden, und andere Immunsuppressiva (z. B. Methotrexat, Azathioprin, Mycophenolatmofetil). Bei geringer Krankheitsaktivität reicht oft der Einsatz von nichtsteroidalen Antiphlogistika, Antimalariamitteln (z. B. Hydroxychloroquin, Chloroquin) oder bisweilen niedrigdosierten Kortikosteroiden aus. Massiver Organbefall erfordert hohe Kortikosteroiddosen (z. B. Prednison 1 mg/kg/Tag) und zusätzlich Immunsuppressiva. Bei Symptomen einer Myositis oder Sklerodermie entspricht das therapeutische Vorgehen diesen Krankheiten.
Patienten mit Raynaud-Syndrom sollten aufgrund ihrer Symptome behandelt werden und -soweit von ihrem Blutdruck toleriert- mit Kalziumkanalblockern (z. B. Nifedipin) und Phosphodiesterase-Hemmern (z. B. Tadalafil).
Alle Patienten sollten engmaschig auf Atheroklerose kontrolliert werden. Patienten mit langfristiger Kortikosteroidtherapie sollten eine Osteoporoseprophylaxe erhalten. Wenn eine immunsuppressive Kombinationstherapie eingesetzt wird, sollten die Patienten eine Prophylaxe für opportunistische Infektionen, wie Pneumocystis jirovecii (siehe Prävention von Pneumocystis jirovecii-Pneumonie), und Impfstoffe gegen häufige Infektionen (z. B. Streptokokkenpneumonie, Influenza, COVID-19) erhalten.
Einige Experten ermutigen zu einem Screening auf Lungenhochdruck in regelmäßigen Abständen mit Lungenfunktionstests und/oder Echokardiographie alle 1 bis 2 Jahre, abhängig von den Symptomen.
Wichtige Punkte
Die MCTD ähnelt sehr oft einem systemischen Lupus erythematodes, einer systemischen Sklerose und/oder Polymyositis.
Typischerweise sind ANA und Antikörper gegen U1 RNP vorhanden und Anti-Sm- und Anti-DNA-Antikörper fehlen, aber das Vorhandensein von Anti-RNP-Antikörpern ist nicht ausreichend, um die Diagnose zu stellen.
Einer pulmonalen Hypertonie ist vorzugreifen.
Die Behandlung milder Krankheitsverläufe erfolgt mit nichtsteroidalem Antiphlogistikum oder Antimalariamitteln und die schwerer Krankheitsverläufe mit Kortikosteroiden und auch anderen Immunsuppressiva.