


Das Charcot-Marie-Tooth-Hoffmann-Syndrom ist eine hereditäre Neuropathie, bei der die Unterschenkelmuskulatur immer schwächer wird und schließlich schwindet (Atrophie).
Das Charcot-Marie-Tooth-Hoffmann-Syndrom betrifft Nerven, die die Muskelbewegung kontrollieren und die sensorischen Informationen an das Gehirn übermitteln.
Die Schwäche setzt im Unterschenkel ein und breitet sich nach und nach über die Gliedmaßen aus. Die Betroffenen verlieren die Fähigkeit zur Wahrnehmung von Vibrationen, Schmerzen und Temperatur.
Zur Absicherung der Diagnose werden eine Elektromyografie und Messungen der Nervenleitungsgeschwindigkeit durchgeführt.
Das Fortschreiten der Erkrankung kann nicht aufgehalten werden, der Einsatz von Schienen, sowie Physio- und Ergotherapie können jedoch die Funktionsfähigkeit verbessern.
(Siehe auch Übersicht über das periphere Nervensystem.)
Das Charcot-Marie-Tooth-Hoffmann-Syndrom ist die häufigste hereditäre Neuropathie; sie betrifft etwa 1 von 2500 Personen. Sie kann in der Kindheit oder später im Leben beginnen.
Das Charcot-Marie-Tooth-Hoffmann-Syndrom ist eine sensorische und motorische Neuropathie. Das bedeutet, dass die motorischen Nerven (zuständig für die Steuerung der Muskelbewegungen) sowie die sensorischen Nerven (die sensorische Informationen ans Gehirn weiterleiten) betroffen sind.
Es gibt verschiedene Arten des Charcot-Marie-Tooth-Hoffmann-Syndroms. Typischerweise wird die Erkrankung jedoch folgendermaßen nach dem Schaden eingestuft, den sie verursacht:
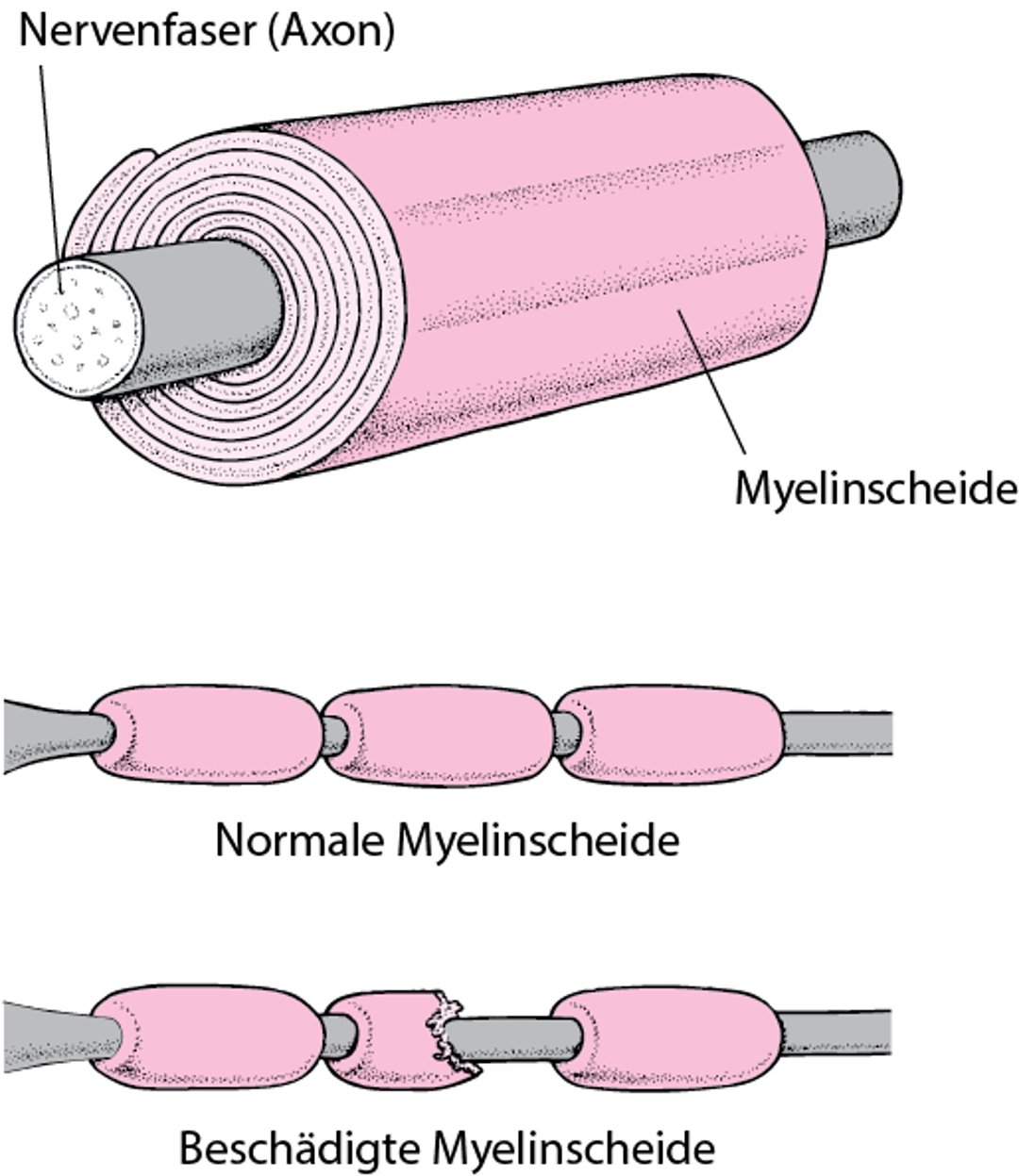
Demyelinisation (Schädigung der Myelinscheide): Die Axone (der Teil der Nervenzellen, der die Signale weiterleitet) leben weiter, aber die Myelinscheide rund um das Axon wird geschädigt oder zerstört (Demyelinisierung). (Die Myelinscheide funktioniert ähnlich wie die Isolierung elektrischer Kabel und ermöglicht die rasche Übermittlung von Nervenimpulsen.)
Schaden am Axon: Hauptsächlich ist das Axon (der Teil der Nervenzellen, der die Signale weiterleitet) betroffen. Bisweilen sterben die Axone ab.
Isolierung einer Nervenfaser
Die meisten Nervenfasern sind von einem vielschichtigen fetthaltigen Mantel (Lipoprotein) umgeben, der Myelinschicht. Diese Schichten bilden die Myelinscheide. Ähnlich der Isolierung eines Elektrokabels ermöglicht die Myelinscheide, Nervensignale (elektrische Impulse) schnell und exakt entlang der Nervenfaser weiterzuleiten. Ist die Myelinscheide defekt (was als Demyelinisation bezeichnet wird), leiten die Nerven Impulse nicht richtig weiter. |

Die meisten Typen des Charcot-Marie-Tooth-Hoffmann-Syndroms werden in der Regel autosomal-dominant (nicht vom Geschlecht abhängig) vererbt. Das bedeutet, dass die Krankheit auch auftritt, wenn das entsprechende Gen vom anderen Elternteil gesund ist. Einige Formen können jedoch rezessiv (d. h. es sind zwei Gene erforderlich, eines von jedem Elternteil) oder auch geschlechts-(X)-gebunden vererbt werden. Bei der geschlechtsgebundenen Vererbung liegt das Gen auf dem X-Chromosom. Dieses Chromosom bestimmt, ob eine Person männlich oder weiblich ist. Männer verfügen über ein X-Chromosom (von ihrer Mutter) und ein Y-Chromosom (von ihrem Vater). Frauen haben zwei X-Chromosomen (eines von der Mutter und eines vom Vater). Wenn ein Mann ein X-Chromosom mit dem veränderten Gen erbt, entwickelt sich die Krankheit. Wenn eine Frau ein verändertes X-Chromosom erbt, wird sie die Krankheit wahrscheinlich nicht entwickeln, da sie auch ein normales (unverändertes) X-Chromosom geerbt hat.
Die Déjerine-Sottas-Krankheit (hypertrophe interstitielle Neuropathie) ist eine seltene erbliche sensorische und motorische Neuropathie. Sie verursacht ähnliche Symptome wie das Charcot-Marie-Tooth-Hoffmann-Syndrom. Allerdings verschlimmert sich die Schwäche weitaus schneller. Es tritt außerdem ein Verlust der Sensorik und der Reflexe ein. Sie beginnt im Kindesalter. Sie kann autosomal-dominant oder -rezessiv (autosomal = unabhängig vom Geschlecht) vererbt werden. Bei der dominanten Form ist nur ein Gen von einem Elternteil erforderlich, damit es zu der Erkrankung kommt, während bei der rezessiven Form zwei Gene, von jedem Elternteil je eines, erforderlich sind.
Symptome des Charcot-Marie-Tooth-Hoffmann-Syndroms
Die Symptome variieren je nach Art des Charcot-Marie-Tooth-Hoffmann-Syndroms.
Bei der einen Art können die ersten Symptome im mittleren Kindesalter oder in der Jugend auftreten. Die Schwäche manifestiert sich zuerst an den Unterschenkeln. Dabei kann man den Knöchel nicht mehr beugen, um den vorderen Teil des Fußes anzuheben (Fallfuß), wobei die Wadenmuskeln verkümmern. Anschließend verkümmert die Handmuskulatur. Es kommt zum Verlust der Positions-, Vibrations-, Schmerz- und Temperaturempfindung in den Händen und Füßen; dieser Verlust schreitet allmählich nach oben in den Gliedmaßen fort.
Bei leichteren Typen der Erkrankung sind Hammerzehen und Hohlfuß unter Umständen die einzigen Symptome. Bei einem Typ haben Männer schwere Symptome, während die Symptome bei Frauen nur leicht oder gar nicht vorhanden sind.
Die Krankheit schreitet langsam voran und beeinträchtigt die Lebenserwartung nicht.
Diagnose des Charcot-Marie-Tooth-Hoffmann-Syndroms
Untersuchung durch den Arzt
Elektromyografie und Messung der Nervenleitfähigkeit
Die Diagnose des Charcot-Marie-Tooth-Hoffmann-Syndroms kann durch folgende Fragen unterstützt werden:
Welche Bereiche des Körpers geschwächt sind
Wann die Erkrankung eingesetzt hat
Ob Familienangehörige ähnliche Symptome haben
Auch wird überprüft, ob die Patienten Fußdeformationen haben (Hohlfuß und Hammerzehe). Diese Informationen können dazu beitragen, die verschiedenen Typen des Charcot-Marie-Tooth-Hoffmann-Syndroms voneinander und von anderen Neuropathien zu unterscheiden.
Zur Absicherung der Diagnose werden eine Elektromyografie und Messungen der Nervenleitungsgeschwindigkeit durchgeführt.
Es stehen Gentests und Beratung beim Charcot-Marie-Tooth-Hoffmann-Syndrom zur Verfügung.
Behandlung des Charcot-Marie-Tooth-Hoffmann-Syndroms
Schienen bei Fallfuß
Manchmal Physio- und Ergotherapie
Es gibt keine Behandlung, die das Fortschreiten des Charcot-Marie-Tooth-Hoffmann-Syndroms aufhält.
Mit Schienen kann die Spitzfußstellung korrigiert werden, manchmal ist eine orthopädische Operation zur Stabilisierung des Fußes erforderlich.
Oft sind Physio- (zur Muskelkräftigung) und Ergotherapie sinnvoll. Eine berufsbegleitende Beratung kann den Betroffenen helfen, ihren Beruf trotz des Fortschreitens der Krankheit weiter auszuüben.